CARACTERIZAÇÃO ESTRUTURAL DOS TRANSPORTADORES ABC DE MOLIBDATO E OLIGOPEPTÍDEOS EM MYCOBACTERIUM TUBERCULOSIS USANDO BIOINFORMÁTICA
STRUCTURAL CHARACTERIZATION OF MOLYBDATE AND OLIGOPEPTIDE ABC TRANSPORTERS IN MYCOBACTERIUM TUBERCULOSIS USING BIOINFORMATICS
LIMA, Bruna Évelyn Briotto1; ZANUNI, Luisa Stenico1; CREMONESI, Aline Sampaio2;
1Graduanda do Curso de Farmácia - Universidade São Francisco; 2Professora Doutora do Curso de Farmácia da Universidade São Francisco.
brunabriotto@gmail.com
RESUMO. Mycobacterium tuberculosis, patógeno da tuberculose, desafia a saúde pública devido à resistência a antibióticos e alta mortalidade associada, tornando necessária a busca constante por novos antimicrobianos. Um potencial alvo terapêutico são os transportadores ABC de molibdato e oligopeptídeos, uma vez que desempenham papel crucial na sobrevivência da bactéria ao importar esses nutrientes essenciais para o interior celular da mesma, sendo então o objetivo deste trabalho identificar e caracterizar estes transportadores em M. tuberculosis a partir de análises in silico. Foi identificado um sistema completo de transportadores do tipo ABC para cada cada um dos nutrientes estudados, compostos por SBPs, permeases e ATPases, organizados em óperons únicos no genoma. Estruturalmente, SBPs apresentaram regiões de peptídeo sinal indicando funções extracelulares, enquanto permeases mostraram seis regiões transmembranares, sugerindo localização nas membranas. As ATPases foram identificadas pela presença do domínio AAA, característico. As SBPs, ModA e OppA, apresentaram modelos das estruturas tridimensionais conservadas, sendo classificadas como SBP do tipo II e a análise de sequências identificou aminoácidos conservados nos bolsões de ligação das SBPs, sugerindo interações com substratos. Esses resultados destacam potenciais alvos para terapias antimicrobianas, fornecendo dados para investigações futuras quanto ao mecanismo de interação entre as SBPs e os substratos transportados por elas.
Palavras-chave: multirresistência; bioinformática; desenho racional de fármacos; biologia estrutural.
ABSTRACT. Mycobacterium tuberculosis, the tuberculosis pathogen, poses a significant challenge to public health due to antibiotic resistance and associated high mortality, necessitating the constant search for new antimicrobials. One potential therapeutic target is the ABC transporters of molybdate and oligopeptides, as they play a crucial role in the bacterium's survival by importing these essential nutrients into its cell interior. Thus, the aim of this study is to identify and characterize these transporters in M. tuberculosis through in silico analyses. A complete system of ABC transporters was identified for each of the studied nutrients, comprising SBP, permeases, and ATPases, organized in unique operons in the genome. Structurally, SBP exhibited signal peptide regions indicating extracellular functions, while permeases showed six transmembrane regions, suggesting membrane localization. ATPases were identified by the presence of the characteristic AAA domain. The SBP, ModA and OppA, displayed conserved three-dimensional structures, classified as type II SBPs, and sequence analysis identified conserved amino acids in the binding pockets of SBP, suggesting interactions with substrates. These results highlight potential targets for antimicrobial therapies, providing insights for future investigations into the interaction mechanism between SBPs and their transported substrates.
Keywords: multidrug resistance; bioinformatics; rational drug design; structural biology.
INTRODUÇÃO
A Mycobacterium tuberculosis, também conhecida como “bacilo de Koch”, é uma bactéria da família Mycobacteriaceae, que necessita de oxigênio para sobreviver (aeróbia) e se difere da maioria das bactérias Gram positivas devido a presença de ácido micólico em sua parede celular, tornando-a resistente a técnica de coloração de Gram e sendo necessário um método específico denominado Ziehl Nielsen para sua identificação (CHEN et al., 2012; SUBHASREE et al., 2017). Tal bacilo é agente etiológico da tuberculose, doença altamente infecciosa e transmissível que afeta principalmente os pulmões e está diretamente relacionada a fatores sociais, portanto mais presente em países emergentes. Sua propagação ocorre através do ar, ao inalar gotículas contaminadas que são liberadas na tosse, espirros, fala ou respiração (OMS, 2023).
A tuberculose é um grande problema de saúde pública, pois é a segunda principal causa de mortes no mundo, ficando atrás somente do vírus da COVID-19. Sua erradicação até o ano de 2023 é uma das metas estabelecidas nos Objetivos de Desenvolvimento Sustentável (ODS) pela Organização das Nações Unidas (ONU). Somente no ano de 2021, ocorreu um aumento de 3% de casos resistentes à rifampicina - antibiótico de primeira linha no combate à doença (OMS, 2023). Com base nesse cenário, vale ressaltar que a multirresistência apresentada pela M. tuberculosis é extremamente preocupante e desafiadora, pois exige o uso de tratamentos alternativos menos eficazes, caracterizados por maior tempo de duração, custo elevado, alta toxicidade e altas taxas de mortalidade (CAMINERO, 2017).
Desse modo, a busca por novos fármacos que sejam eficientes contra a tuberculose tem como um de seus alvos os transportadores ABC. Essa família de proteínas de transporte atua na exportação de substâncias nocivas, como os antibióticos para as bactérias, e também na importação de nutrientes essenciais para o funcionamento da célula, sendo fundamentais no desenvolvimento e sobrevivência de diversas cepas farmacorresistentes. Do ponto de vista estrutural, os transportadores ABC geralmente consistem em dois domínios transmembrana (Transmembrane Segments - TMS’s) formando um canal de passagem, dois domínios de ligação de nucleotídeos (Nucleotide Binding Domains - NBP’s) fornecendo energia ao processo e uma proteína ligadora de substrato (Substrate Binding Protein - SBP) que se conecta de modo específico a molécula que será transportada (OLIVEIRA; BALAN, 2020).
O molibdato, íon oxigenado derivado do molibdênio, é um dos nutrientes importados pelos transportadores ABC que desempenha um papel fundamental como cofator enzimático em diversas reações bioquímicas (YANG et al., 2021). Por sua vez, os oligopeptídeos, compostos orgânicos formados por cadeias de ligações peptídicas constituídas principalmente por aminoácidos, também são importados pelo sistema ABC de transporte, permitindo a adaptação da bactéria ao ambiente através do fornecimento de nitrogênio e aminoácidos por fontes adicionais (LIMA et al., 2014). Sabendo-se da importância desses nutrientes e dos transportadores ABC para desenvolvimento da bactéria e considerando a atual escassez de medicamentos para combater as cepas multirresistentes, o presente trabalho teve como objetivo identificar e caracterizar as proteínas que compõem os transportadores ABC de fosfato a partir de análises in silico, a fim de contribuir futuramente para um futuro design de antimicrobianos com alvo nos transportadores ABC de molibdato e oligopeptídeos realmente eficazes.
METODOLOGIA
Identificação do transportador ABC de molibdato e oligopeptídeos em Mycobacterium tuberculosis
A identificação das proteínas constituintes do transportador ABC de molibdato e oligopeptídeos da M. tuberculosis foi realizada utilizando o banco de dados online KEGG (Kyoto Encyclopedia of Genes and Genomas - https://www.genome.jp/kegg/ - KANEHISA et al., 2007), no qual também foi feita a identificação da sequência assinatura ABC a partir da sequência de aminoácidos das proteínas indicadas como ATPases. A indicação da região promotora, identificada a partir da sequência de nucleotídeos de ambas as proteínas, foi efetuada pelo uso da plataforma Softberry BPROM (http://www.softberry.com/ - SALAMOVE; SOLOVYEVAND, 2011)
Classificação das proteínas
As proteínas SBP’s foram identificadas pelo programa Signal P 6.0 devido a presença de peptídeo sinal, aminoácidos geralmente presentes na região N-terminal da proteína que a direcionam ao seu local de atuação (https://services.healthtech.dtu.dk/services/SignalP-6.0/ - TEUFEL et al., 2022). As proteínas permeases dos transportadores foram reconhecidas pela identificação de domínios transmembrana realizada pelo software TMHMM (Transmembrane Hidden Markov Model - http://www.cbs.dtu.dk/services/TMHMM/ - KROGH et al., 2001), que utiliza modelos ocultos de Markov para detectar padrões de hélices transmembrana presentes na sequência de aminoácidos fornecida (PATRA et al.; 2020). Outro domínio altamente conservado identificado através do programa SMART domain (Architecture Research Tool) é o AAA+, característico de proteínas ATPase (PUCHADES; SANDATE; LANDER, 2020). O software FoldIndex forneceu a previsão das regiões desordenadas das proteínas a partir da sua sequência de aminoácidos (https://fold.proteopedia.org/cgi-bin/findex - PRILUSKY et al., 2005). Para a identificação das estruturas secundárias das proteínas classificadas como SBPs utilizou-se o programa PsiPred (http://bioinf.cs.ucl.ac.uk/psipred/ - MCGUFFIN et al., 1999).
Modelagem tridimensional e alinhamento de aminoácidos
O modelo tridimensional da proteína alvo foi determinado através da modelagem ab initio que visa calcular estruturas moleculares a partir da mecânica quântica, e por homologia, assim utilizando uma proteína de estrutura conhecida e altamente compatível como molde (WEBB; SALI, 2016). O programa utilizado foi o ColabFold que, a partir da sequência de aminoácidos da proteína projeta sua estrutura tridimensional, método que avalia a qualidade da mesma, devido a fornecer um conjunto de ângulos de torção que são formados (https://toolkit.tuebingen.mpg.de/tools/hhpred - SOUZA et al., 2021). Para a visualização, análise do modelo, previsão do sítio de ligação e estimativa da afinidade de interação com os ligantes, foi utilizado o programa PyMol (https://pymol.org/2/. - DELANO, 2002). Com o intuito de analisar a conservação de aminoácidos possivelmente envolvidos na ligação de molibdato e oligopeptídeos foram alinhadas as sequências de aminoácidos das proteínas homólogas no programa Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/ - LARKIN et al., 2007). A proteína similar utilizada na comparação dos aminoácidos no PyMol foi identificada através do programa BLAST (Basic Local Alignment Search Tool - https://blast.ncbi.nlm.nih.gov/Blast.cgi - ALTSCHUL et al., 1997).
RESULTADOS E DISCUSSÃO
Mycobacterium tuberculosis possui transportadores do tipo de ABC completos para captação e transporte de Molibdato e Oligopeptídeos
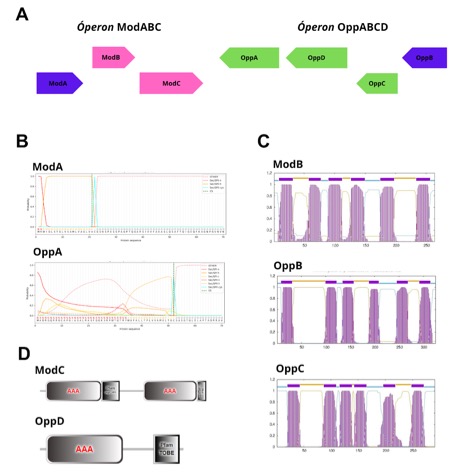
A partir do banco de dados KEGG, foi identificado um sistema de transporte do tipo ABC para molibdato e outro para oligopeptídeos em M. tuberculosis. Os transportadores de molibdato são compostos por três proteínas denominadas ModA, ModB e ModC, classificadas, respectivamente, como SBP, TMD e NBD. Por sua vez, foram identificadas quatro proteínas envolvidas no transporte de oligopeptídeos em M. tuberculosis: uma transportadora de substrato (OppA), duas permeases (OppB e OppC) e uma ATPase (OppD). Realizada a análise do mapa genômico, verificou-se que cada um dos sistemas está organizado em um único óperon (Figura 1A). Tal organização possibilita a ativação ou desativação dos genes como uma unidade funcional, simplificando o processo de transcrição realizado pela bactéria, já que são regulados pela mesma região promotora (MENON, 2021).
A região promotora é fundamental na transcrição dos genes, pois sinaliza onde a RNA polimerase deve iniciar esse processo (ZAHA; FERREIRA; PASSAGLIA, 2014), sendo uma informação importante também para compreender a regulação gênica, apresentando um potencial significativo para o desenvolvimento de estratégias de modulação como alvo terapêutico de fármacos. Desse modo, as análises das proteínas responsáveis pela captação de substratos (ModA e OppA), utilizando a plataforma softberry BPROM e a sequência de nucleotídeos à montante (upstream), identificaram a região promotora na área intergênica, iniciando em -10 e terminando em -35, conforme previsto em estudos anteriores em procariotos (MENON, 2021). A partir disso, caracteriza-se como promotores as proteínas ModA e OppB, já que elas estão localizadas no início do óperon.
As classificações das proteínas obtidas no KEGG foram confirmadas por meio de programas específicos. A provável região de peptídeo sinal foi localizada pelo programa Signal P 6.0, na região N-terminal das proteínas ModA e OppA, sendo previsto estar localizado entre os aminoácidos 1 a 21 e entre 1 e 52, respectivamente (Figura 1B). As regiões de peptídeo sinal funcionam como guias para destinos específicos (ARMENTEROS, 2019), sugerindo que a proteína desempenha funções fora do citoplasma (SILVA, 2020), funcionando provavelmente como SBP's.
O programa TMHMM identificou seis regiões transmembranas nas proteínas ModB, OppB e OppC, sugerindo que são proteínas do tipo permease (Figura 1C). É possível identificar essas regiões devido às características estruturais dessas proteínas, que são adaptadas para interagir com as membranas celulares, portanto são compostas principalmente por aminoácidos hidrofóbicos e estruturas secundárias para facilitar o transporte do substrato através da membrana (GUNA, 2018). As proteínas ATPases são dependentes da energia gerada a partir da hidrólise de ATP, devido a isso possuem sequências singulares conservadas que são encarregadas de realizar tais atividades celulares (DEVLIN, 2011). As sequências de aminoácidos das proteínas ModC e OppD analisadas no SMART domain revelaram a presença do domínio AAA+ comum em proteínas ATPases (Figura 1D). Além disso, foi identificada a região P-loop, caracterizada por um loop alfa-beta-Rossman que conserva aminoácidos associados à assinatura ABC, e as regiões Walker A e B (PUCHADES; SANDATE; LANDER, 2019). Essas regiões são formadas por sequências de aminoácidos conservados que caracterizam as proteínas do tipo ATPases como pertencentes ao sistema de transporte ABC.
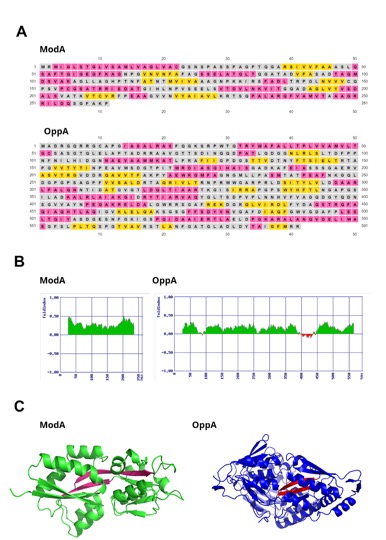
Para a realização de métodos experimentais é essencial identificar previamente se as proteínas estudadas são estáveis. Portanto, o programa FoldIndex permitiu a identificação de porções desordenadas a partir da hidrofobicidade e carga elétrica da proteína (PRILUSKY, et al., 2005), gerando gráficos que indicaram a ausência de regiões propensas a serem desordenadas na ModA, enquanto OppA apresentou duas pequenas regiões que possivelmente serão desordenadas (Figura 2A). Desse modo, pode-se dizer que ambas as proteínas seriam de fácil manipulação in silico. Em ambas as proteínas os resultados obtidos na análise do PsiPred sugeriram um número expressivo de coils (regiões em espiral ou desordenadas), uma quantidade relevante de Alfa-Hélices e poucas Fitas Betas (Figura 2B), perfil esperado em proteínas do tipo SBP (LOCHER, 2016). Tal análise se baseia em padrões de organização da cadeia polipeptídica de cada uma das proteínas, predizendo sua estrutura secundária (BUCHAN; JONES, 2019).
A modelagem da estrutura tridimensional das proteínas transportadoras de substrato, o programa ColabFold, que é uma implementação adaptada do software AlphaFold (MIRDITA et al., 2022), gerou 5 modelos de cada uma das proteínas SBP, sendo eles ranqueados em ordem crescente de confiabilidade. Os modelos gerados e classificados em primeiro lugar foram analisados no Pymol. Comumente as proteínas SBP’s apresentam dois domínios globulares interligados por uma região de dobradiça, que confere flexibilidade ou rigidez essencial às mesmas. Desse modo, há uma classificação baseada nas características dessa região de conexão, sendo proteínas tipo I aquelas cuja conexão entre os domínios é estabelecida por uma cadeia polipeptídica, proteínas tipo II possuem duas fitas-beta como elemento conector, enquanto as do tipo III apresentam uma alfa-hélice desempenhando esse papel (RAHMAN; MACHUCA; ROUJEINIKOVA, 2021). A análise dos modelos gerados demonstra que ambas as proteínas apresentam duas fitas-beta entre os dois domínios globulares, característico de proteínas transmembrana tipo II (Figura 2C).
Figura 1 – Caracterização dos transportadores ABC de Molibdato e Oligopeptídeos. A. Representação esquemática dos óperons que estão possivelmente envolvidos na captação de molibdato e oligopeptídeo da bactéria M. tuberculosis, sendo ModA e OppB os respectivos promotores (representados em azul), indicando a direção em que se inicia a transcrição (representada pelas setas); B. Análise do peptídeo sinal das proteínas ligadoras de substrato pelo programa SignalIP, sendo possível observar a identificação do mesmo em ModA entre os aminoácidos 1 e 22 e em OppA de 1 a 52. C. Análise do domínio transmembrana dado pelo programa TMHMM na qual a cor roxa representa a região transmembrana das proteínas, a linha azul representa os aminoácidos citoplasmáticos e a linha laranja as regiões externas à membrana; D. Esquema representando a presença das sequências de domínio AAA+, sugerindo assim que as proteínas ModC e OppD são proteínas ATPases.
Fonte: Elaborado pelos autores.
As proteínas ligadoras de substratos apresentam conservação estrutural da classe e aminoácidos responsáveis pela interação com ligantes.
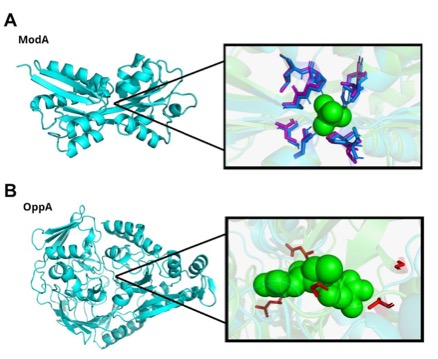
O alinhamento múltiplo de sequências de proteínas SBP de transportadores de molibdato das bactérias Escherichia coli, Xanthomonas citri e Mycobacterium tuberculosis realizado no Clustal permitiu identificar aminoácidos conservados, que possivelmente estão voltados para o bolsão de ligação dessas proteínas e interagem com o nutriente (Figura 3A). Para analisar estruturalmente essa conservação da ModA de M. tuberculosis, foi identificada no BLAST a proteína de maior identidade: ModA (PDB ID: 1AMF - HU et al., 1997) de Escherichia coli e realizado o alinhamento estrutural no PyMol. A análise revelou similaridades estruturais entre ambas as proteínas, uma vez que a preservação da estrutura ancestral é crucial para a manutenção da função proteica (FILHO; ALENCASTRO, 2003). Além disso, foram identificados os aminoácidos A²⁴, A²⁵, A²⁶, S²⁷, L²⁸, S⁵⁵, S⁷³, V¹³², P¹³³, V¹⁷⁴ e Y¹⁷⁵ que são conservados e constituem o bolsão de ligação (Figura 4A). Tais aminoácidos têm cadeias laterais não polares, dificultando a interação com água. Sendo assim, tendem a se agruparem no interior da proteína, contribuindo na estabilidade, formação de núcleos hidrofóbicos e interação com o ligante (BARREIRO; FRAGA, 2014). De acordo com estudos realizados anteriormente, o alinhamento múltiplo de sequências de 15 ortólogos de ModA de diferentes bactérias apresentou esses mesmos aminoácidos como possíveis regiões conservadas (MOUTRAN, 2009). O estudo não abrangeu a M. tuberculosis, porém os resultados obtidos nas presentes análises convergem com os anteriores, direcionando os resultados para a importância de tais aminoácidos.
A identificação de aminoácidos conservados no bolsão de proteínas SBPs de transportadores ABC tem sido uma importante estratégia para identificar e caracterizar interações entre SBPs e diferentes moléculas. Estudos envolvendo a análise dos aminoácidos e as características do bolso de interação das SBPs ligadoras de poliaminas trouxeram esclarecimentos definidos dos mecanismos de interação deste tipo de transportador (SILVA; FIGUEIREDO; CREMONESI, 2023) bem como adaptações para otimizar a captação de nutrientes para a célula (CREMONESI et al., 2021)
As mesmas análises foram realizadas com OppA. O alinhamento múltiplo de sequências de proteínas SBP de transportadores de oligopeptídeos foi realizado comparando as proteínas das mesmas bactérias (Figura 3B), permitindo a identificação de aminoácidos conservados cuja probabilidade de constituírem o bolsão de ligação foi analisada no alinhamento do PyMol, utilizando a proteína similar pertencente a Brucella melitensis (PDB ID: 4ONY - artigo a ser publicado). Na comparação entre essas proteínas não foi possível identificar aminoácidos conservados voltados ao bolsão de ligação. Esse resultado provavelmente está relacionado à baixa porcentagem de identidade entre as proteínas alinhadas. Contudo, foram identificados os aminoácidos G⁵⁷, A⁶⁰, L²⁹², V⁴²⁹, G⁴⁴¹, N⁴⁵⁹ e G⁵³⁵ na proteína modelada que, em estudos anteriores, foram indicados como conservados e constituintes do bolsão de ligação (Figura 4B) (CHAI et al., 2022).
Figura 2 – Representação da estrutura secundária e análise da ordenação das proteínas. A. Gráfico que representa a estrutura secundária das proteínas ModA e OppA respectivamente, no qual a cor rosa indica as Hélices- alfa, a cor cinza indica as regiões de coil - torções ao redor da proteína e a cor amarela indicando as Fitas-betas; B. Análise que representa a ordenação das proteínas, onde as partes representadas na cor verde são suas partes ordenadas e as representadas em vermelho suas áreas de desordem; C. Modelagem das proteínas ModA e OppA, indicando as Fitas-Beta na cor rosa e vermelho, feito a partir do programa AlphaFold.
Fonte: Elaborado pelos autores.
Figura 3 – Representação dos resultados do alinhamento múltiplo de SBP de transportadores ABC de molibdato e oligopeptídeos A. Alinhamento de sequência das proteínas transportadoras de molibdato de E. coli, X. citri e M. tuberculosis, em vermelho estão representados os aminoácidos conservados e em cinza suas devidas posições. B. Alinhamento de sequência das proteínas transportadoras de oligopeptídeos de E. coli, X. citri pv e M. tuberculosis, em vermelho estão representados os aminoácidos conservados e em cinza suas devidas posições.

Fonte: Elaborado pelos autores.
Figura 4 – Alinhamento estrutural de aminoácidos conservados. A. A proteína ModA está representada à esquerda e os aminoácidos conservados estão representados na ampliação da região do bolsão de aminoácidos à direita. A cor azul representa os aminoácidos da proteína similar e roxo os pertencentes a ModA. Ambos estão situados ao redor do molibdato (representado na cor verde) e voltados para o bolsão de ligação. B. Os aminoácidos conservados estão localizados no bolsão de ligação, que está ampliado, e são representados pela cor vermelha, estando posicionados ao redor do oligopeptídeo, retratado pela cor verde.
Fonte: Elaborado pelos autores.
CONCLUSÃO
A partir dos resultados obtidos neste estudo, é possível sugerir que dentre os transportadores ABC de M. tuberculosis, existe um responsável pela captação e importação de molibdato para o interior da célula bacteriana e outro pela captação e importação de oligopeptídeos, nutrientes essenciais para a bactéria. Esses transportadores são compostos por proteínas SBP (ModA e OppA), TMD (ModB, OppB e OppC) e NBD (ModC e OppD). Os genes que codificam essas proteínas estão organizados em um único óperon em ambos os casos. As proteínas ModA e OppA modeladas se mostraram altamente conservadas com relação às outras SBP’s descritas na literatura. Todas são constituídas por dois domínios globulares conectados por duas fitas beta, além de manterem aminoácidos conservados no bolsão de ligação relacionados à interação com o ligante. Visualizada as estruturas tridimensionais das proteínas, sugere-se que há onze aminoácidos conservados presentes na região do bolsão possivelmente interagem com a molécula de molibdato e sete aminoácidos que possivelmente interagem com oligopeptídeos. Para a confirmação dessas interações ainda são necessários mais estudos. Os dados obtidos podem ser utilizados para melhor entendimento dos mecanismos de ação dos transportadores ABC e para a identificação de novas moléculas, a partir da docagem molecular, que sejam potenciais alvos de fármacos por realizarem a interação proteína-ligante impedindo a captação de molibdato e oligopeptídeos para a M. tuberculosis.
REFERÊNCIAS
ALTSCHUL, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research, v. 25, p. 3389-3402, 1997. DOI: https://doi.org/10.1093/nar/25.17.3389
ARMENTEROS, J. J. A.; TSIRIGOS, K.; SONDERBY, C. K.; PETERSEN, T. N.; WINTHER, O.; BRUNA K., S.; HEIJNE, G. v.; NIELSEN, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, v. 37, n. 4, p.420-423, 2019. DOI: https://doi.org/10.1038/s41587-019-0036-z
BARREIRO, E J.; FRAGA, C. A. M. Química Medicinal: As bases moleculares da ação dos fármacos. Artmed Editora, 2014.
BUCHAN, D. W. A., JONES, D. T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic acids research, v.2, n. 47, 2019. (W1), W402–W407. DOI: https://doi.org/10.1093/nar/gkz297
CAMINERO, L. JA. Challenges and Outlooks in Multi-drug Resistant Tuberculosis. Arch Bronconeumol. 2017 Aug. v.53, n. 8, p.417-418. English, Spanish. DOI: https://doi.org/10.1016/j.arbres.2017.01.018
CHAI, H.; KIM, Y.; HAM, J.; KIM, T.; LIM, D. Identifying ligand-binding specificity of the oligopeptide receptor OppA from Bifidobacterium longum KACC91563 by Structure-based molecular modeling. Research Square; 2020. DOI: https://doi.org/10.1016/j.arabjc.2022.104198
CHEN, Ping et al. A highly efficient Ziehl-Neelsen stain: identifying de novo intracellular Mycobacterium tuberculosis and improving detection of extracellular M. tuberculosis in cerebrospinal fluid. Journal of clinical microbiology, v. 50, n. 4, p. 1166-1170, 2012. DOI: https://doi.org/10.1128/jcm.05756-11
Cremonesi, A.S.; De la Torre, L.I.; Degenhardt, M.F.S.; Muniz,G.S.V.; Lamy, V.T.; Oliveira, C.L.P. BALAN, A. The citrus plant pathogen Xanthomonas citri has a dual polyamine-binding protein. Archives of Biochemistry and Biophysics, v. 28, p. 1-12, 2021. DOI: https://doi.org/10.1016/j.bbrep.2021.101171
DELANO, W. L. The PyMol Molecular Graphics System DeLano Scientific, 2002. http://www.pymol.org/
DEVLIN, T. M. Manual de bioquímica: com correlações clínicas. Editora Blucher, 2011.
FILHO, O. A. S.; ALENCASTRO, R. B. Modelagem de proteínas por homologia. Química Nova, v. 26, p. 253-259, 2003. DOI: https://doi.org/10.1590/S0100-40422003000200019
GUNA, A.; HEDGE, R. S. Transmembrane Domain Recognition during Membrane Protein Biogenesis and Quality Control. Current Biology Review, v.28, 2018. DOI: https://doi.org/10.1016/j.cub.2018.02.004
HU, Y., RECH, S., GUNSALUS, R. et al. Crystal structure of the molybdate binding protein ModA. Nat Struct Mol Biol, v.4, p.703–707, 1997. DOI: https://doi.org/10.1038/nsb0997-703
KANEHISA, M. et al. KEGG for linking genomes to life and the environment. Nucleic acids research, v. 36, n. suppl_1, 2007, p. D480-D484. DOI: https://doi.org/10.1093/nar/gkm882
KROGH, A. et al. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journal of Molecular Biology, v. 305, n. 3, p. 567-580, 2001. DOI: https://doi.org/10.1006/jmbi.2000.4315
LARKIN, M. A. et al. Clustal W and Clustal X version 2.0. bioinformatics, v. 23, n. 21, p. 2947-2948, 2007. DOI: https://doi.org/10.1093/bioinformatics/btm404
LIMA, R. N. S.; COSTA, S. O. P. D.; FERREIRA, R. C. C. O Transporte de Oligopeptídeos na fisiologia e patogênese de bactérias do gênero Streptococcus. Revista de Microbiologia, Janeiro, 2014.
LOCHER, K. P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol, v.23, p. 487-493, 2016. DOI: https://doi.org/10.1038/nsmb.3216
MCGUFFIN, L. J.; BRYSON, K.; JONES, D. T. The PSIPRED protein structure prediction server. Bioinformatics Applications Note, v. 16, p. 404-405, 2000. DOI: https://doi.org/10.1093/bioinformatics/16.4.404
MENON, S.; PIRAMANAYAKAM, S.; AGARWAL, G. Computational identification of promoter regions in prokaryotes and Eukaryotes. EPRA International Journal of Agriculture and Rural Economic Research (ARER), v. 9, n. 7, p. 21-28, 2021. DOI: https://doi.org/10.36713/epra7667
MIRDITA, M., SCHUTZE, K., MORIWAKI, Y. et al. ColabFold: making protein folding accessible to all. Nat Methods,v.19, p.679–682, 2022. DOI: https://doi.org/10.1038/s41592-022-01488-1
MOUTRAN, A. Modelagem molecular das proteínas captadoras de Molibdato (ModA) e Oligopeptídeo (OppA) de Xanthomonas axonopodis pv. citri. 2009. Tese (Doutorado em Microbiologia) – Universidade de São Paulo, São Paulo, 2009. DOI: https://doi.org/10.11606/T.42.2009.tde-16072009-100344
OLIVEIRA, M. C. B.; BALAN, A. The ATP-Binding Cassette (ABC) transport systems in Mycobacterium tuberculosis: Structure, function, and possible targets for therapeutics. Biology, v. 9, n. 12, p. 443, 2020. DOI: https://doi.org/10.3390/biology9120443
ORGANIZAÇÃO MUNDIAL DA SAÚDE. Relatório Global sobre Tuberculose. Genebra: OMS, 2023. ISBN: 978-92-4-008385-1
PATRA, P. et al. Epitope-based vaccine designing of nocardia asteroides targeting the virulence factor mce-family protein by immunoinformatics approach. International Journal of Peptide Research and Therapeutics, v. 26, p. 1165-1176, 2020. DOI: https://doi.org/10.1007/s10989-019-09921-4
PRILUSKY, J., FELDER, C. E., ZEEV-BRN-MORDEHAI, T., RYDBERG, E. H., MAN, O., BECKMANN, J. S., SILMAN, I., SUSSMAN, J. L. FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics (Oxford, England), v. 21, n. 16, p. 3435–3438, 2005. DOI: https://doi.org/10.1093/bioinformatics/bti537
PUCHADES, C.; SANDATE, C. R.; LANDER, G. C. The molecular principles governing the activity and functional diversity of AAA+ proteins. Nature Reviews Molecular Cell Biology, v. 21, n. 1, p. 43-58, 2020. DOI: https://doi.org/10.1038/s41580-019-0183-6
RAHMAN, M. M.; MACHUCA, M. A.; ROUJEINIKOVA, A. Bioinformatics analysis and biochemical characterisation of ABC transporter-associated periplasmic substrate-binding proteins ModA and MetQ from Helicobacter pylori strain SS1. Biophysical Chemistry, v. 272, p. 106577, 2021. DOI: https://doi.org/10.1016/j.bpc.2021.106577
SALAMOV, V. S. A.; SOLOVYEVAND, A. Automatic annotation of microbial genomes and metagenomic sequences. Metagenomics and its applications in agriculture, biomedicine and environmental studies, p. 61-78, 2011.
SILVA, G. A. A. Caracterização do antígeno proteico ssaa de Staphylococcus saprophyticus utilizando estratégias in silico e modelo ex vivo de infecção. 2020. Tese (Mestrado em Genética e Biologia Molecular) - Universidade Federal de Goiás, Goiás 2020. http://repositorio.bc.ufg.br/tede/handle/tede/10597
SILVA, K.M.; FIGUEIREDO, N.G.; CREMONESI, A.S. Use of Bioinformatics Techniques in the Characterization of Genes and Proteins Involved in the Transport of Polyamines from Staphylococcus Genus. JSM Bioinformatics, Genomics and Proteomics, v. 6, n.1, 2023. DOI: https://doi.org/10.47739/2576-1102/1041
SOUZA, C. et al. Estratégia Algorítmica para a Reconstrução e Validação da Estrutura Molecular de Variantes do SARS-CoV-2. Anais do XV Brazilian e-Science Workshop. SBC, 2021. p. 65-72. DOI: https://doi.org/10.5753/bresci.2021.15790
SUBHASREE, C. R. et al. Review on comparative genomics for mycobacterium tuberculosis strains. International Journal of Pharmaceutical Sciences and Research, v. 8, n. 12, p. 5022-5042, 2017. DOI: https://doi.org/10.13040/IJPSR.0975-8232.8(12).5022-42
TEUFEL, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nature biotechnology, v. 40, n. 7, p. 1023-1025, 2022. DOI: https://doi.org/10.1038/s41587-021-01156-3
WEBB, B.; SALI, A. Comparative protein structure modeling using MODELLER. Current protocols in bioinformatics, v. 54, n. 1, 2016. DOI: https://doi.org/10.1002/cpbi.3
YANG X, LIU H, ZHANG Y, SHEN X. Roles of Type VI Secretion System in Transport of Metal Ions. Front Microbiol., v.5, n.1, 2021. DOI: https://doi.org/10.3389/fmicb.2021.756136
ZAHA, A.; FERREIRA, H. B.; PASSAGLIA, L.M.P. Biologia Molecular Básica-5. Artmed Editora, 2014.
Recebido em: 01/04/2024.
Publicado em: 28/08/2024.