USO DE TECNOLOGIA DE BARREIRAS PARA PROCESSOS ASSÉPTICOS NA ÁREA FARMACÊUTICA
USE OF BARRIER TECHNOLOGIES FOR ASEPTIC PROCESS IN THE PHARMACEUTICAL AREA
DOI:10.24933/e-usf.v9i1.466
v.9n.1 (2025)
PAIVA, Bárbara Damario1; SIMÕES, Luciana Taba Nakazato1; PINTO, Marcio Cardoso2
1Alunas do Curso de Farmácia da Universidade São Francisco
2 Professor Doutor do Curso de Farmácia da Universidade São Francisco
RESUMO. A tecnologia de barreira desempenha um papel indispensável na produção de produtos estéreis na indústria farmacêutica, sendo um importante parâmetro para assegurar a qualidade, eficácia e segurança dos medicamentos. Essa tecnologia inclui sistemas de isolamento físico, como isoladores e sistemas de barreiras de acesso restrito, que minimizam o risco de contaminação cruzada e exposição a agentes externos durante os processos de fabricação e envase. No Brasil, a Agência Nacional de Vigilância Sanitária (ANVISA) regulamenta a produção de medicamentos estéreis. Segundo essa regulamentação, as operações em áreas classificadas devem ser realizadas sob condições controladas, com monitoramento contínuo de parâmetros ambientais, como partículas, microrganismos e fluxo de ar. Considerando a importância do assunto, este estudo teve como objetivo apresentar um referencial teórico sobre o uso de tecnologia de barreiras para processos assépticos na área farmacêutica. O desenvolvimento metodológico envolveu uma revisão narrativa da literatura. Com base nos achados ficou evidente que a adoção da tecnologia de barreira deve atender às exigências normativas a fim de reduzir a necessidade de intervenções manuais em áreas críticas, melhorando a proteção do produto e a segurança do operador. A implementação de sistemas de barreira possibilita maior controle sobre os processos assépticos. Além disso, a tecnologia contribui para o cumprimento de padrões internacionais, alinhando a produção nacional às exigências do mercado global.
Palavras-chave: tecnologia de barreira; processos assépticos; produtos estéreis; isoladores.
ABSTRACT. Barrier technology plays an indispensable role in the production of sterile products in the pharmaceutical industry, being an important parameter to ensure the quality, efficacy and safety of drugs. This technology includes physical isolation systems, such as isolators and restricted access barrier systems, which minimize the risk of cross-contamination and exposure to external agents during the manufacturing and packaging processes. In Brazil, the National Health Surveillance Agency (ANVISA) regulates the production of sterile drugs. According to this regulation, operations in classified areas must be carried out under controlled conditions, with continuous monitoring of environmental parameters, such as particles, microorganisms and air flow. Considering the importance of the subject, this study aimed to present a theoretical framework on the use of barrier technology for aseptic processes in the pharmaceutical area. The methodological development involved a narrative review of the literature. Based on the findings, it was evident that the adoption of barrier technology must meet the regulatory requirements in order to reduce the need for manual interventions in critical areas, improving product protection and operator safety. The implementation of barrier systems enables greater control over aseptic processes. In addition, the technology contributes to compliance with international standards, aligning national production with the demands of the global market.
Keywords: barrier technology; aseptic processes; sterile products; isolators.
INTRODUÇÃO
Medicamentos estéreis são produtos farmacêuticos que devem estar completamente livres de microrganismos uma vez que são usados em situações em que a contaminação microbiana pode representar risco à saúde do paciente, como injeções, infusões intravenosas, oftálmicos, implantes e medicamentos utilizados em procedimentos cirúrgicos. A produção de medicamentos estéreis deve seguir critérios rigorosos de controle ambiental, técnicas assépticas e processos de esterilização para garantir sua qualidade, segurança e eficácia. A esterilidade é considerada um requisito crítico e está regulamentada por normas nacionais e internacionais. No Brasil, a indústria farmacêutica deve atender as regulamentações definidas pela Agencia Nacional de Vigilância Sanitária (ANVISA) (BRASIL, 2022).
Dada relevância do tema, os autores Cunha, Baiense e Andrade (2023), discutem sobre o papel das salas limpas na garantia da qualidade de produtos estéreis e injetáveis na indústria farmacêutica e descrevem que as salas limpas são ambientes projetados para controlar níveis de contaminação por partículas e microrganismos e devem atender aos padrões de qualidade exigidos nesse setor. Os autores discutem os principais parâmetros que influenciam a eficácia das salas limpas, como a classificação ambiental, controle de partículas, fluxo de ar unidirecional, pressão positiva, e sistemas de filtragem HEPA (High-Efficiency Particulate Air). Além disso, enfatizam a importância do treinamento de profissionais, uso de vestimentas adequadas e protocolos de limpeza para minimizar riscos de contaminação (CUNHA; BAIENSE; ANDRADE, 2023).
A manufatura e controle de qualidade de medicamentos estéreis requerem boas práticas que minimizem os riscos de contaminação, seja microbiológica ou por partículas totais. A contaminação microbiana pode provocar alterações organolépticas e comprometer a eficácia terapêutica (LIRIO, 2019). Em se tratando de partículas totais no ambiente, de acordo com o Guia da Indústria Food and Drug Administration (FDA, 2004), seu controle é significativo, pois podem estar presentes no produto como contaminantes estranhos ou até mesmo atuar como carreador de microrganismos.
As seguintes medidas de prevenção de contaminação devem ser consideradas: as instalações, equipamentos e processos devem ser desenhados, qualificados e validados de acordo com os guias de boas práticas de fabricação; os operadores devem ser treinados e devidamente qualificados; as matérias-primas e materiais de embalagem devem ser provenientes de fornecedores qualificados, e devem ser testados e aprovados antes do uso (EUROPEAN COMMISSION, 2022). O Guia FDA (2004) também aborda a minimização da exposição de artigos estéreis aos potenciais riscos de contaminação, indicando um elevado grau de controle ambiental, com equipamentos que ofereçam qualidade de ar ISO 5 e otimização dos fluxos de processo como pontos essenciais para atingir um alto padrão de garantia de esterilidade.
A qualidade de ar ISO 5 (International Organization for Standardization) refere-se a um nível de pureza do ambiente controlado conforme a classificação da ISO 14644-1, que regula salas limpas e áreas controladas. Essa classificação determina a quantidade máxima permitida de partículas suspensas no ar por metro cúbico, garantindo condições ideais para processos que exigem alta esterilidade, como a fabricação de medicamentos estéreis (ISO, 2015). No que se refere a fabricação de medicamentos estéreis, de acordo com a Instrução Normativa (IN) n° 35/2019 da Anvisa (BRASIL, 2019), são distinguidos quatro graus de limpeza:
o Grau A: zona para operações de alto risco (ex: zona de envase, onde são feitas manipulações e conexões assépticas), normalmente em estação de trabalho com fluxo de ar unidirecional ou isolador.
o Grau B: o ambiente circundante da área Grau A.
o Graus C e D: áreas limpas para realização de etapas menos críticas da fabricação de medicamentos estéreis.
Conforme o European Commission (2022), a avaliação do nível de contaminação microbiana é parte da qualificação da área limpa. Tanto na qualificação, como no monitoramento ambiental durante o processo, nenhum crescimento microbiano é permitido em um ambiente classificado como Grau A.
A norma ISO 14644-1 (2015) especifica a classificação do ar com base na concentração de partículas entre 0,1µm e 5,0µm em salas e zonas limpas. Estas salas ou zonas são espaços em que a concentração de partículas em suspensão no ar é controlada e são desenhadas, construídas e operadas de maneira a controlar a introdução, geração e retenção de partículas no seu interior. A concentração máxima permitida de partículas no ar para cada grau está descrita na Tabela 1.
Tabela 1. Concentração máxima permitida de partículas no ar.
|
Grau |
Número Máximo Permitido de Partículas/m3 de Ar |
|||
|
Em repouso |
Em Operação |
|||
|
≥ 0,5µm |
≥ 5,0µm |
≥ 0,5µm |
≥ 5,0µm |
|
|
A |
3.520 |
20 |
3.520 |
20 |
|
B |
3.520 |
29 |
352.000 |
2.900 |
|
C |
352.000 |
2.900 |
3.520.000 |
29.000 |
|
D |
3.520.000 |
29.000 |
Não definido |
Não definido |
Fonte: BRASIL (2019).
O uso de tecnologia de barreira para aumentar a proteção do produto de potenciais fontes de contaminação deve ser considerada em substituição das tradicionais áreas limpas. Os isoladores, por exemplo, promovem uma barreira mecânica separando o ambiente interno estéril da área externa ao redor, e podem ser abertos ou fechados, mas devem garantir condição de ambiente classificado como Grau A onde o produto estiver exposto (EUROPEAN COMMISSION, 2022). Podem ser construídos com parede flexível (exemplo: PVC - policloreto de vinila) (DREMOVA et al., 2023) ou com parede rígida composta de aço inox e/ou vidro (FDA, 2004).
O uso de sistemas de barreiras de acesso restrito ou RABs (sigla do termo em inglês designado por Restricted Access Barrier Systems) constitui uma alternativa ao uso de isoladores devido a sua flexibilidade, podendo operar de modo fechado (closed RABs) ou aberto (open RABs). No modelo closed RABs, o equipamento permanece fechado desde a descontaminação até a operação. Enquanto o modelo aberto permanece com a barreira fechada na maior parte do tempo, mas tem a possibilidade do operador abrir as portas para realizar intervenções pré-definidas, de acordo com o International Society for Pharmaceutical Engineering (ISPE) (LYSFJORD, 2005).
Diante das considerações sobre a importância da esterilidade na manufatura de medicamentos estéreis, o objetivo deste estudo consistiu em realizar um levantamento teórico sobre o uso de tecnologia de barreiras para processos assépticos na área farmacêutica em áreas críticas Grau A e avaliar a minimização do risco de contaminação.
METODOLOGIA
O artigo foi elaborado por meio de uma revisão narrativa de literatura. Esta trata-se de um estudo que busca reunir, sintetizar e interpretar informações já publicadas sobre um tema, sem o rigor metodológico de revisões sistemáticas. É frequentemente utilizada para explorar conceitos amplos, contextualizar pesquisas ou descrever o estado atual do conhecimento em um campo de estudo e permite que o autor desenvolva uma abordagem descritiva e crítica sobre o tema (CORDEIRO, et al., 2007). Para este fim, foram analisadas as informações e dados em artigos científicos, de língua portuguesa e inglesa publicados na base de dados Pubmed, Google acadêmico, Scielo, sites de revistas e jornais acadêmicos, acerca do tema, publicados no período de 2019 a 2024. Adicionalmente foram analisados guias, normas, livros, farmacopeias e relatórios publicados pela ANVISA, Parenteral Drug Association (PDA), International Society for Pharmaceutical Engineering (ISPE), International Organization for Standardization (ISSO); International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), Food and Drug Administration (FDA), United States Pharmacopeia (USP) e European Comission, publicados entre 2001 e 2024.
Para busca de dados foram utilizadas palavras-chave como “tecnologias de barreiras para processos assépticos”, “isoladores”, “RABs”, “produção de medicamentos estéreis”. Para a exclusão e inclusão de informações foram utilizados como parâmetro a leitura de introdução, ano de publicação e idioma português e inglês.
Figura 1. Fluxograma da metodologia
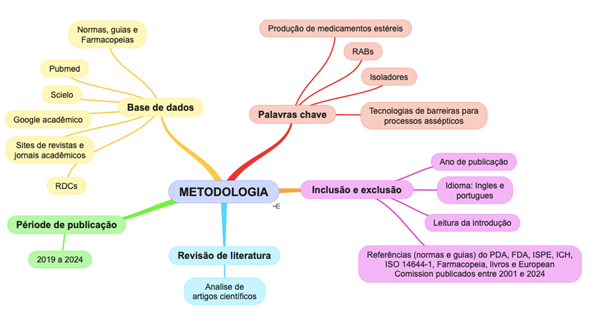
Fonte: Próprio autor.
RESULTADOS E DISCUSSÃO
A manufatura de produtos estéreis envolve um conjunto de processos para garantir a segurança e eficácia dos medicamentos, e deve ser aplicado o Gerenciamento de Riscos da Qualidade (GRQ), ou Quality Risk Management (QRM), conforme artigo técnico publicado em 2024 pelo Instituto de Pesquisas Tecnológicas (IPT), sobre Fabricação de Medicamentos Estéreis. Esse gerenciamento deve incluir um projeto adequado das instalações, dos equipamentos e dos processos, contando com um programa robusto para monitorar a adequada implementação e seu respectivo desempenho.
De acordo com o Guia da ANVISA sobre Gerenciamento de Riscos da Qualidade (BRASIL, 2023), que é baseado no guia ICH Q9(1) (2023), a fabricação de um medicamento e seus componentes contam com um grau de risco, inclusive o risco à sua qualidade. E a qualidade do produto é garantida a partir de tomada de decisões apropriadas e baseadas em riscos ao longo do ciclo de vida do produto.
Adicionalmente, a normativa europeia European Commission (2022) já exige a chamada Estratégia de Controle de Contaminação (Contamination Control Strategy – CCS), que se refere a um documento que descreve de forma holística a abordagem para garantir a qualidade do produto, definindo todas as questões críticas associadas ao processo, avaliando a eficácia de medidas de controle e monitorando dos mecanismos utilizados para gerenciar os riscos de qualidade e segurança do produto. O CCS deve ser continuamente revisto e atualizado, como forma de melhoria contínua dos métodos de manufatura e controle para minimizar os riscos de contaminação por microrganismos, endotoxinas e partículas visíveis e sub-visíveis. Este documento deve abranger tópicos tais como: desenho da planta produtiva e do processo de manufatura, equipamentos, pessoas, utilidades, controle de matéria-prima e de material de embalagem, qualificação de fornecedores, gerenciamento dos prestadores de serviços, validação de processos, manutenção preventiva, limpeza e desinfecção, sistemas de monitoramento ambiental, gerenciamento de mudanças, investigações de não-conformidades e respectivas ações preventivas e ações corretivas. A verificação da efetividade deste controle deve ser parte de uma revisão gerencial periódica.
Os seguintes procedimentos para prevenir contaminação devem ser considerados para os processos assépticos: salas limpas controladas e monitoradas com frequência, projetadas com a escolha correta de materiais, considerando superfícies lisas e não porosas, fáceis de limpar e que não liberam partículas (CUNHA; BAIENSE; ANDRADE, 2023); controle de temperatura e umidade do ambiente; fornecimento de ar filtrado através de filtros de alta eficiência sob pressão positiva; sistema de monitoramento de condições ambientais; sistema de limpeza e desinfecção de sala e equipamentos e um sistema de manutenção dos equipamentos utilizados para o controle das condições assépticas (FDA, 2024).
Em relação a paramentação, esta deve ser apropriada para o processo e utilizada de forma a proteger o produto da contaminação. Os operadores de área grau A/B devem utilizar vestimentas limpas, estéreis ou adequadamente sanitizadas a cada sessão de trabalho. As luvas devem ser desinfetadas regularmente, as máscaras e luvas devem ser substituídas no mínimo a cada sessão de trabalho. E devem ser usados capuzes que envolvam todo o cabelo e, quando aplicável, protetor para barba e bigode (BRASIL, 2019).
As superfícies, o ar e as pessoas devem ser monitorados após operações críticas, sendo que os resultados devem ser considerados ao se revisar a documentação referente ao lote para a liberação do produto acabado, que devem atender o nível de contaminação microbiana, conforme descrito na Tabela 2.
Tabela 2 – Limites recomendados para contaminação microbiana (valores médios)
|
Grau |
Amostra volumétrica ativa (UFC/m3) |
Placas de 90mm (UFC/4horas)
|
Placas de contato 55mm (UFC/placa) |
Luvas 5 dedos (UFC/luva) |
|
A |
<1 |
<1 |
<1 |
< 1 |
|
B |
10 |
5 |
5 |
5 |
|
C |
100 |
50 |
25 |
- |
|
D |
200 |
100 |
50 |
- |
Fonte: BRASIL, 2019.
A aplicação de tecnologias adequadas na fabricação e controle de medicamentos, como os isoladores e RABs, pode reduzir os riscos de contaminação. De acordo com o Parenteral Drug Association (PDA) (2001), os isoladores são capazes de manter o ambiente livre de microrganismos detectáveis por “isolar” a zona crítica de trabalho dos operadores humanos, filtrando o ar de entrada e descontaminando a estação de trabalho, bem como os materiais nela usados, utilizando-se vapores, tais como peróxido de hidrogênio ou ácido peracético, permitindo que todas as superfícies expostas sejam atingidas durante o processo de descontaminação. O método de descontaminação tem que ser automatizado, qualificado e aprovado.
O isolador deve ser qualificado antes do uso, passando pelas qualificações de instalação, operação e de performance. A qualificação de instalação consiste numa descrição detalhada de aspectos físicos do sistema, como dimensão, configuração interna e material de construção. A qualificação de operação inclui a checagem de alarmes, teste de integridade com manutenção da pressão durante a operação, desenvolvimento do ciclo de descontaminação com verificação de distribuição do agente responsável pela descontaminação do equipamento, inclusive com a sua carga cheia (USP, 2024). Na qualificação de performance, o uso de indicadores químicos e bioindicadores é necessário, sendo que estes devem estar bem distribuídos no interior da câmara para confirmar a eficácia da descontaminação. Para os bioindicadores, uma redução de pelo menos 106 esporos/indicador é esperada (PDA, 2001).
O desenho de um isolador de modelo aberto deve garantir condição de ambiente Grau A com proteção de primeiro ar nas zonas críticas e fluxo de ar unidirecional, enquanto o isolador de modelo fechado deve garantir condição de ambiente Grau A com proteção adequada ao produto exposto durante o processo. O ar não precisa ser unidirecional quando operações simples são conduzidas, no entanto o fluxo turbulento não deve oferecer risco de contaminação ao produto exposto. Para os isoladores abertos a área adjacente deve ser classificada como Grau C e para os fechados Grau D. Para isso deve avaliar os riscos previamente, considerando aspectos como um programa de bio-descontaminação, a extensão da automação, o impacto das manipulações das luvas e da perda da integridade da barreira/luva (EUROPEAN COMMISSION, 2022). A Figura 2 ilustra a classificação dos ambientes em que se localizam os isoladores e RABs.
Figura 2 – Esquema da classificação ambiental conforme preconizado pelo European Comission (2022)
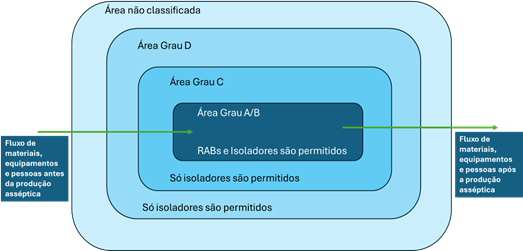
Fonte: TANZINI et al., 2023.
Os isoladores de parede flexível transparente, conforme ilustrado na Figura 3A, como os confeccionados com PVC, são usados para obter um ambiente estéril ou controlado em termos microbiológicos, proporcionando em geral uma estação de trabalho mais ampla e com maior visibilidade (DREMOVA et al., 2023). Os isoladores de parede rígida, indicada na Figura 3B, são mais resistentes a agentes de limpeza e promovem melhor contenção (PDA, 2001).
Figura 3. (A) Isolador para Teste de Esterilidade (parede flexível); (B) Isolador para Teste de Esterilidade (parede rígida).

Fonte: Acervo da ANTIBIÓTICOS DO BRASIL LTDA, 2024.
Existe também um outro tipo de isolador, o modelo SA25, que se refere a um sistema robótico totalmente fechado, sem conexões para luvas e geralmente utilizado para enchimento asséptico de frascos e seringas (Figura 4). O SA25 elimina a intervenção por operadores na zona crítica e suprime falhas como as de vazamento de luvas, promovendo uma importante melhoria na garantia de esterilidade na produção de medicamentos injetáveis (McCALL et al., 2022).
Figura 4. Isolador modelo SA25.

Fonte: McCALL et al., 2022.
Os RABs, sistemas de barreiras de acesso restrito (Figuras 6 A e B), têm o papel de evitar os riscos de contaminação, podendo se apresentar nos modelos ativo ou passivo. O RABs ativo possui filtros HEPA e unidades de ventilação no interior da barreira, enquanto o modelo passivo utiliza os filtros HEPA da própria sala limpa (ELSAID; PAZHAYATTI, 2024). Os RABs devem possuir um ambiente adjacente classificado no mínimo como Grau B, logo é fundamental a realização de estudos do fluxo de ar para confirmar a ausência do ingresso de ar na câmara proveniente do ambiente adjacente durante as intervenções (EUROPEAN COMMISSION, 2022).
Figura 6. (A) Sistema de Barreira de Acesso Restrito (RABs); (B) Sistema de barreira de acesso restrito (RABS).

Fonte: (A) Acervo da ANTIBIÓTICOS DO BRASIL LTDA, 2024; (B) LUCHI et al., 2024.
As principais diferenças entre os isoladores dos RABs podem ser observadas na Tabela 3.
Tabela 3 – Diferenças entre Isoladores e RABs
|
|
Isoladores |
RABs |
|
Desenho |
Aberto: Garantir ambiente Grau A com fluxo unidirecional.
Fechado: Garantir ambiente Grau A com fluxo de ar unidirecional ou turbulento.
Pressão negativa somente quando necessária a contenção do produto (exemplo: radiofármacos). |
Garantir ambiente Grau A com fluxo de ar unidirecional e proteção de primeiro ar na zona crítica.
Fluxo de ar positivo da zona crítica para a área adjacente. |
|
Área adjacente |
Aberto: Mínimo Grau C Fechado: Mínimo Grau D
|
Mínimo Grau B |
|
Luvas |
O teste de integridade é realizado geralmente no início e ao final de cada lote, ou campanha produtiva, ou sessão de produção. A descontaminação ocorre junto com a estação de trabalho.
|
Devem ser esterilizadas antes da instalação e esterilizadas ou descontaminadas por método validado antes de cada campanha produtiva. Se expostas ao ambiente adjacente uma desinfecção deve ser realizada. |
|
Descontaminação |
Processo automatizado, validado e controlado com parâmetros de ciclos pré-definidos utilizando agentes esporicidas na forma gasosa ou de vapor, a fim de obter uma zona crítica livre de microrganismos viáveis. |
Aplicação de agente esporicida por método validado e que demonstre que todas as áreas internas estão apropriadas para o processo asséptico. |
Fonte: EUROPEAN COMISSION, 2022.
A inovação nas áreas de produtos biotecnológicos e nanotecnologia de medicamentos vem recebendo destaque recentemente, principalmente devido a pandemia do COVID-19. Antes de 2015 somente 13 nanomedicamentos haviam sido aprovados pelo FDA. Em 2021, 100 nanomedicamentos estavam sendo comercializados e 563 estavam em fase de estudo clínico ou outro estágio de desenvolvimento. As áreas terapêuticas envolvem tratamento de câncer (53%) e infecções (14%), assim como outras doenças endócrinas, do sistema nervoso, imunológico, cardiovascular, ocular e pele, além das estratégias inovadoras de vacinação baseadas nos nanomedicamentos. As mudanças na regulamentação, principalmente no que está relacionado nos processos assépticos, e o uso de sistemas robóticos no ambiente farmacêutico para atender os requisitos de Boas Práticas de Fabricação vem sendo observada. Apesar da indústria farmacêutica ser caracterizada, devido a aspectos regulatórios, por aderir de forma mais lenta o uso de novas tecnologias, recentemente o European Medicines Agency (EMA) liderou o incentivo da melhoria dos processos utilizando tecnologias já estabelecidas em outros segmentos de manufatura.
O Anexo 1 do Guia das Boas Práticas de Fabricação (EudraLex, Volume 4, Parte I), referente as orientações sobre a fabricação de medicamentos estéreis, foi revisto e engloba alterações relacionadas com as condições ambientais no âmbito da fabricação de medicamentos estéreis. A nova versão do Anexo 1 publicada pela Comissão Europeia (EUROPEAN COMISSION, 2022) introduziu a visão inovadora das indústrias farmacêuticas e forneceu ferramentas de melhorias de processo usando a robótica, visando a prevenção de contaminação na produção de medicamentos estéreis, mas também dos não-estéreis. As palavras-chave “contaminação” e “risco” aparecem respectivamente 116 e 100 vezes na edição de 2022 do Anexo I. Já na versão anterior em 2008, apareceram 35 e 20 vezes, respectivamente. O uso de salas limpas convencionais era uma escolha aceitável na edição de 2008. Já a edição de 2022 endereça o uso de tecnologia de barreiras como preferencial. Associada a esta mudança, os conceitos de automação e robótica são introduzidos para diminuir o risco de contaminação e exposição de pessoas. Particularmente num ambiente asséptico, o uso de automação e robôs torna o seguimento dos procedimentos operacionais mais efetivo, e reduz os riscos de segurança e de falha humana. Além do que, permite uma eficaz rastreabilidade das operações, com mais geração de dados de parâmetros críticos de processo, permitindo uma melhor análise de tendência e identificação de não-conformidades. Desta forma, ações corretivas e preventivas podem ser tomadas mais rapidamente, limitando o risco de perda do controle da qualidade de produção (TANZINI et al., 2023).
A utilização de sistemas de isoladores se estende para outras finalidades além da indústria farmacêutica, como por exemplo, em estudos biomédicos com animais livres de contaminação microbiana. A produção destes animais gnotobióticos, que possuem microbiota associada definida, deve ser realizada em ambientes dotados de barreiras sanitárias absolutas (ANDRADE; PINTO; OLIVEIRA, 2002). Na revisão realizada por Dremova et al. (2023), o uso do isolador segrega efetivamente camundongos estéreis dos microrganismos presentes no ambiente externo, bem como permite a colonização destas espécies com uma cultura de microrganismos pré-determinada. Devido a segurança e toxicidade, agentes a base de cloro são geralmente utilizados para a descontaminação, com eficácia contra bactérias e esporos. O conhecimento técnico e o treinamento da equipe são fundamentais, pois a falha de um único parâmetro, como a introdução de um material não esterilizado adequadamente, um dano na barreira física do isolador, uma ruptura nas luvas ou nas vedações do equipamento, pode comprometer a esterilidade do isolador e dos animais.
Com o aumento da prevalência de pacientes com câncer, o uso de sistemas robotizados em cabines com tecnologia de barreira para a preparação de medicamentos citotóxicos em farmácias hospitalares tem aumentado, a fim de minimizar a contaminação das áreas de trabalho, uma vez que há uma carga operacional aumentada e um estresse ocupacional relacionados ao preparo de quimioterápicos. Quando recebe a prescrição, o agente quimioterápico é reconhecido e confirmado, possibilitando a preparação por um braço robótico de acordo com o algoritmo de manufatura. Uma revisão sistemática realizada por Shin et al. (2023) concluiu que o uso de preparação por sistema robótico tem mais acurácia do que o uso de um sistema manual, reduzindo o risco de contaminação na área de preparação, nas embalagens de infusão e nas luvas.
CONCLUSÃO
Este estudo teve como objetivo realizar um levantamento teórico sobre o uso de tecnologia de barreiras para processos assépticos na área farmacêutica em áreas críticas Grau A, avaliando a minimização do risco de contaminação. Com base na contextualização realizada, foi possível concluir que a produção de medicamentos estéreis segue critérios rigorosos de controle ambiental, técnicas assépticas e processos de esterilização para garantir a qualidade, segurança e eficácia. A esterilidade é considerada um requisito crítico e os Sistemas de Barreiras de Acesso Restrito, são tecnologias utilizadas em ambientes controlados, como salas limpas na indústria farmacêutica, para proteger produtos estéreis durante sua fabricação e envase. Esses sistemas criam uma barreira física entre o operador e o ambiente crítico, reduzindo significativamente o risco de contaminação microbiológica e por partículas. Nos últimos tempos, o incentivo do uso de tecnologia de barreiras e sistemas automatizados tem aumentado na indústria farmacêutica, principalmente com o objetivo de reduzir do risco de contaminação durante a manufatura e controle de qualidade de medicamentos. Com a publicação do Anexo I, da European Comission em 2022, os fabricantes de medicamentos estéreis vêm aprimorando a avaliação e adequação dos processos, definindo estratégias de controle de contaminação com base na Gestão de Riscos da Qualidade.
A implementação de tecnologias de barreira resulta em ambientes mais robustos para a fabricação de produtos estéreis, assegurando o cumprimento dos rigorosos requisitos de qualidade exigidos para medicamentos injetáveis e outras formas farmacêuticas críticas. Além disso, essas tecnologias são consideradas relevantes em um cenário de crescente complexidade nas formulações e maior exigência regulatória, garantindo não apenas a conformidade, mas também a sustentabilidade dos processos de fabricação. Investir em sistemas de barreiras é, portanto, uma estratégia indispensável para indústrias farmacêuticas que buscam excelência operacional e confiança no mercado global.
REFERÊNCIAS
ANDRADE, A.; PINTO, S. C.; OLIVEIRA, R. S. Animais de Laboratório: Criação e Experimentação. Rio de Janeiro: Editora FIOCRUZ, 2002. 388 p. ISBN: 85-7541-015-6. Disponível em: <cap_08_animais_labor.pmd>. Acesso em 08 nov. 2024.
BRASIL. Agência Nacional de Vigilância Sanitária. Resolução RDC nº 658, de 30 de março de 2022 dispõe sobre as Diretrizes Gerais de Boas Práticas de Fabricação de Medicamento. Brasília, 2022.
BRASIL. Agência Nacional de Vigilância Sanitária. Resolução Instrução Normativa n.o 35 de 21 de agosto de 2019. Dispõe sobre as Boas Práticas Complementares a Medicamentos Estéreis. Disponível em: < bf4ab3da-b130-43ab-913d-243fd2efb86f> Acesso em 06 set. 2024.
BRASIL. Agência Nacional de Vigilância Sanitária. Guia de Gerenciamento de Riscos da Qualidade no 62 de 19 de julho de 2023. Disponível em: < 4bda3e21-c404-4e18-9783-0ff6da5f548e (anvisa.gov.br)>. Acesso em 26 set. 2024.
CORDEIRO, Alexander Magno et al. Revisão sistemática: uma revisão narrativa. Revista do colégio brasileiro de cirurgiões, v. 34, p. 428-431, 2007.
CUNHA, R. S.; BAIENSE, A. S. R.; ANDRADE, L. G. Salas limpas na indústria farmacêutica: garantindo a qualidade de produtos estéreis e injetáveis. Revista ibero-americana de humanidades, ciências e educação-REASE. São Paulo, v.9.n.11. nov. 2023. Disponível em: <https://periodicorease.pro.br/rease/article/view/12606/5919>. Acesso em: 27 set. 2024.
DREMOVA, O.; MIMMLER, M.; PAESLACK, N.; KHUU, M. P.; Gao Z.; BOSMANN, M.; GARO, L. P.; SCHÖN, N.; MECHLER, A.; BENEICH, Y.; REBLING, V.; MANN, A.; PONTAROLLO, G.; KIOUPTSI, K.; REINHARDT, C. Sterility testing of germ-free mouse colonies. Front Immunol. 2023. Disponível em: < https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10662041/>. Acesso em: 23 ago. 2024.
ELSAID, A; PAZHAYATTIL, A. Legacy Filling Lines Evolve Safeguarding with RABS Technology. PDA Letter, 2024. Disponivel em: https://www.researchgate.net/profile/Ajay-B-Pazhayattil/publication/380168613_Legacy_Filling_Lines_Evolve_Safeguarding_with_RABS_Technology/links/662f423206ea3d0b74167ef2/Legacy-Filling-Lines-Evolve-Safeguarding-with-RABS-Technology.pdf. Acesso em: 24 out. 2024.
EUROPEAN COMMISSION. The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex I – Manufacture of Sterile Mecinal Products. 2022. Disponível em: <https://health.ec.europa.eu/system/files/2022-08/20220825_gmp-an1_en_0.pdf>. Acesso em 23 ago. 2024.
ICH. International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. ICH Q9 - Quality Risk Management, 2006a. Disponível em: < https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmoni-sation-technical-requirements-registration-pharmaceuticals-human-use_en-3.pdf >. Acesso em: 15 out. 2024.
ISO. International Organization for Standardization. ISO 14644-1 - Cleanrooms and associated controlled environments – Part 1: Classification of air cleanliness by particle concentration. 2015.
FDA. Food and Drug Administration. 21 CFR part 211. Current Good Manufacturing Practice for Finished Pharmaceuticals. 2024. Disponível em: <eCFR :: 21 CFR Part 211 -- Current Good Manufacturing Practice for Finished Pharmaceuticals>. Acesso em 19 set. 2024.
FDA. Food and Drug Administration. Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice. 2004. Disponível em: <https://www.fda.gov/media/71026/download>. Acesso em 23 ago. 2024.
LIRIO, A. C. M. Abordagem microbiológica e estatística aplicada ao monitoramento ambiental em áreas produtivas farmacêuticas. 2019. 132p. Dissertação (Mestrado). Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, 2019.
LUCHI, A; RODRIGUES, M. F. A; COUVO, N. S; LÉO, P. Fabricação de Medicamentos Estéreis: Diretrizes para Controle de Contaminação. Revista IPT - Tecnologia e Inovação v.8, n.25, abr., 2024. Disponível em: <Fabricação de Medicamentos Estéreis: Diretrizes para Controle de Contaminação | Revista IPT: Tecnologia e Inovação>. Acesso em: 19 set. 2024.
LYSFJORD, J. ISPE Definition: Restricted Access Barrier Systems (RABS) for Aseptic Processing. 2005. Pharmaceutical Engineering. Disponível em: <ISPE Definition: Restricted Access Barrier Systems (RABS) for Aseptic Processing (a3p.org)>. Acesso em: 19 set. 2024.
MCCALL, J; BARNARD, N; GADIENT, K; KASIREDDY, C; KURTZ, A; LI, Y; PAGE, T; PUTNAM, T; BRENNAN, Á. Environmental Monitoring for Closed Robotic Workcells Used in Aseptic Processing: Data to Support Advanced Environmental Monitoring Strategies. AAPS PharmSciTech. 2022 Aug 3;23(6):215. doi: 10.1208/s12249-022-02360-3. Erratum in: AAPS PharmSciTech. 2022 Aug 30;23(7):245. doi: 10.1208/s12249-022-02398-3. PMID: 35922577. Disponível em: <https://pubmed.ncbi.nlm.nih.gov/35922577/>. Acesso em: 18 out. 2024.
PDA. Parenteral Drug Association. Technical Report n. 34: Design and Validation of Isolators Systems for the Manufacturing and Testing of Health Care Products. 2001. PDA Journal of Pharmaceutical Science and Technology. Vol. 55. n. 5.
SHIN, S.; KOO, J.; KIM, S. W.; KIM, S.; HONG, S. Y.; LEE, E. Evaluation of Robotic Systems on Cytotoxic Drug Preparation: A Systematic Review and Meta-Analysis. Medicina (Kaunas). 2023 Feb 22;59(3):431. doi: 10.3390/medicina59030431. PMID: 36984432; PMCID: PMC10056266. Disponível em: <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10056266/>. Acesso em: 18 out. 2024.
TANZINI, A; RUGGERI, M; BIANCHI, E; VALENTINO, C; VIGANI, B; FERRARI, F; ROSSI, S; GIBERTI, H; SANDRI, G. Robotics and aseptic processing in view of regulatory requirements. Pharmaceutics. 2023 May 24;15(6):1581. doi: 10.3390/pharmaceutics15061581. PMID: 37376030; PMCID: PMC10305582. Disponível em: <https://pubmed.ncbi.nlm.nih.gov/37376030/>. Acesso em: 27 set. 2024.
USP. United States Pharmacopeia <1208> Sterility Testing – Validation of Isolators Systems, 2024. Disponível em: <https://online.uspnf.com/uspnf/document/1_GUID-AA521856-522E-4CAD-97B0-A2F2FCD61EA1_2_en-US?source=Activity>. Acesso em: 11 nov. 2024.
Recebido em: 09/12/2024
Publicado em: 29/04/2025