ANÁLISE COMPUTACIONAL DA CAPTAÇÃO DE FOSFATO EM STAPHYLOCOCCUS AUREUS E PROSPECÇÃO DE POTENCIAIS INIBIDORES
Computational Analysis of Phosphate Uptake in Staphylococcus aureus and Screening for Inhibitors
DOI:
CHAVES, Beatriz Nascimento1; SOUSA, Tina Turner Lima de1; CREMONESI, Aline Sampaio2
1Discentes do curso de Biomedicina pela Universidade São Francisco; 2Docente da Universidade São Francisco.
aline.cremonesi@usf.edu.br
RESUMO. Staphylococcus aureus é um patógeno oportunista associado a diversas infecções e à crescente resistência a antibióticos. O fosfato é essencial para processos celulares e, em S. aureus, sua captação ocorre por meio de transportadores do tipo ABC, nos quais a proteína SBP tem papel fundamental. Este estudo teve como objetivo identificar e caracterizar a proteína SBP de S. aureus por meio de abordagens bioinformáticas, visando a identificação de possíveis ligantes capazes de interferir em sua função. Foram utilizados bancos de dados, plataformas online e ferramentas de bioinformática para caracterização estrutural e modelagem da proteína e seus ligantes. A análise revelou um transportador completo, sendo a proteína PstS responsável pela ligação e captação do fosfato. Sua estrutura, típica de proteínas SBP do tipo II, apresenta dois resíduos chave na interação com o fosfato: D116 e T171. A docagem molecular indicou que sulfato, nitrato e diversas moléculas fosfatadas apresentam afinidade com a PstS, com valores negativos de energia livre. Os resultados sugerem que compostos contendo fosfato podem interagir com a PstS, potencialmente interferindo no transporte de fosfato. Esses achados abrem caminho para o desenvolvimento de moléculas que comprometam a captação de fosfato por S. aureus, reduzindo sua viabilidade e potencial infeccioso.
Palavras-chave: Docagem; Bioinformática; Resistência bacteriana.
ABSTRACT. Staphylococcus aureus is an opportunistic pathogen associated with various infections and increasing antibiotic resistance. Phosphate is essential for cellular processes, and in S. aureus, its uptake occurs through ABC-type transporters, in which the SBP protein plays a key role. This study aimed to identify and characterize the SBP protein of S. aureus using bioinformatics approaches to identify potential ligands capable of interfering with its function. Databases, online platforms, and bioinformatics tools were used for structural characterization and molecular modeling of the protein and its ligands. The analysis revealed a complete transporter, with the PstS protein responsible for phosphate binding and uptake. Its structure, characteristic of type II SBP proteins, contains two key residues involved in phosphate interaction: D116 and T171. Molecular docking indicated that sulfate, nitrate, and various phosphate-containing molecules exhibit affinity for PstS, as evidenced by negative free energy values. These findings suggest that phosphate-containing compounds may interact with PstS, potentially disrupting phosphate transport. This study paves the way for the development of molecules that impair phosphate uptake in S. aureus, reducing its viability and infectious potential.
Keywords: Docking; Bioinformatics; Bacterial resistance.
INTRODUÇÃO
Staphylococcus aureus é uma bactéria amplamente disseminada e responsável por inúmeras infecções cutâneas e invasivas graves em todo o mundo. Destaca-se como um dos principais agentes etiológicos de pneumonia, infecções do trato respiratório, infecções associadas a próteses articulares, infecções cardiovasculares e bacteremia hospitalar. Estimativas de 2012 indicam uma incidência anual de 20 a 50 casos de bacteremia por S. aureus a cada 100.000 pessoas, com taxas de mortalidade variando entre 10% e 30% (Cheung et al., 2021). Sua elevada patogenicidade deve-se, em grande parte, à notável capacidade de adaptação a diferentes condições ambientais e ao desenvolvimento de resistência a múltiplos antibióticos. Cepas dessa bactéria têm demonstrado resistência à maioria dos fármacos utilizados no tratamento de infecções causadas por bactérias Gram-positivas, incluindo beta-lactâmicos, glicopeptídeos e oxazolidinonas (Mlynarczyk-Bonikowska et al., 2022). Esse fenômeno representa um grande desafio para a saúde pública global.
A sobrevivência de S. aureus depende da captação de nutrientes essenciais para diversos processos bioquímicos, como metabolismo celular e síntese de DNA (Cassat; Skaar, 2011). Estudos demonstram a relevância dos sistemas de transporte de nutrientes mediados por transportadores do tipo ABC (ATP Binding Cassette – cassete de ligação ao ATP), uma vez que falhas nesse mecanismo comprometem a formação de biofilmes, a resistência bacteriana e a expressão de fatores de virulência (Pereira et al., 2017; Sampaio et al., 2017; Tanaka et al., 2017; Lewis et al., 2012). Esses transportadores, presentes em membranas celulares de todos os organismos, utilizam a energia proveniente da hidrólise de ATP para realizar processos de importação e exportação de moléculas (Tanaka et al., 2017; Ter Beek, Guskov; Slotboom, 2014). Enquanto os transportadores exportadores são encontrados tanto em eucariotos quanto em procariotos, transportando compostos lipídicos e proteicos, os transportadores importadores são exclusivos de procariotos e desempenham papel fundamental na captação de nutrientes essenciais para a célula bacteriana (Locher, 2016; Rees et al., 2009; Ter Beek, Guskov; Slotboom, 2014). Esses sistemas possuem dois domínios ligadores de nucleotídeos (Nucleotide Binding Domains – NBD), responsáveis pelo fornecimento de energia por meio da hidrólise de ATP, além de domínios transmembranares (Transmembrane Domains – TMD), que formam o canal pelo qual os nutrientes são transportados. Nos transportadores importadores, há ainda uma proteína adicional, denominada Substrate Binding Protein (SBP), cuja função é reconhecer e entregar o substrato às TMDs (Lewinson; Livnat-Levanon, 2017).
Dentre os nutrientes essenciais à proliferação de S. aureus, o fosfato desempenha um papel fundamental na síntese de ácidos nucleicos, fosfolipídios e ATP, além de estar envolvido em outros processos metabólicos cruciais (Aguena, 2007). A captação de fosfato inorgânico (Pi) em S. aureus ocorre por meio do sistema de alta afinidade PstSCAB, um transportador da família ABC. Estudos indicam que a deleção desse sistema reduz significativamente a virulência da bactéria, evidenciando a importância da proteína captadora de Pi durante infecções (Kelliher et al., 2020; Pegos et al., 2017).
Diante disso, torna-se essencial aprofundar o conhecimento sobre os mecanismos de captação de fosfato em S. aureus e investigar potenciais inibidores dessa via. O presente estudo tem como objetivo identificar o sistema PstSCAB nessa bactéria e caracterizar a proteína SBP ligadora de fosfato, visando compreender seus mecanismos de interação e explorar possíveis ligantes que possam interferir nesse processo.
METODOLOGIA
Identificação do Óperon
As proteínas transportadoras de fosfato de Staphylococcus aureus foram identificadas a partir de buscas no banco de dados Kegg (Kyoto Encyclopedia of Genes and Genomes – Enciclopédia de Genes e Genomas de Kyoto). As sequências de nucleotídeos e aminoácidos de cada proteína foram analisadas por alinhamento de sequências no Clustal (Larkin et al., 2007), permitindo a comparação dos possíveis aminoácidos envolvidos na ligação do fosfato e a construção da árvore filogenética. As sequências utilizadas para alinhamento foram obtidas no Kegg ou no banco PDB (Protein Data Bank – Banco de Dados de Proteínas) para aquelas proteínas que possuem estrutura resolvida.
A estrutura do óperon foi analisada com base no mapa genômico de S. aureus disponível no Kegg, enquanto a região promotora foi identificada por meio da plataforma Softberry BPROM (Salamov; Solovyevand, 2011), que examina os nucleotídeos à montante do primeiro gene do óperon. O programa BPROM identifica potenciais regiões promotoras em sequências de nucleotídeos, reconhecendo as regiões -35 e -10, que são conservadas em promotores de organismos procariotos (Salamov; Solovyevand, 2011; Reece et al., 2010).
Identificação dos Domínios Formadores do Transportador ABC de Captação de Fosfato
Proteínas secretadas, como as SBPs, necessitam de uma região sinalizadora que direciona seu transporte até o local de atuação. Essa região, denominada peptídeo sinal, geralmente se encontra na porção N-terminal da proteína e é clivada quando esta atinge seu destino (Von Heijne, 1990). Como as análises bioinformáticas são mais precisas quando se utiliza a sequência da proteína funcional, foi necessário identificar e remover o peptídeo sinal antes das análises. Para isso, as sequências de aminoácidos obtidas no banco Kegg foram processadas no programa SignalP 5.0 (Armenteros et al., 2019), permitindo a detecção das regiões a serem removidas.
A predição de regiões ordenadas nas proteínas SBPs foi realizada por meio do programa FoldIndex, que avalia características físico-químicas dos aminoácidos da sequência, identificando possíveis regiões desordenadas (Prilusky et al., 2005). Os domínios funcionais foram determinados com a ferramenta SMART (Simple Modular Architecture Research Tool – Ferramenta de Pesquisa de Arquitetura Modular Simples) (Letunic; Khedkar; Bork, 2021). Além disso, as proteínas permeases do transportador ABC de fosfato foram identificadas com o programa DeepTMHMM (Hallgren et al., 2022), que analisa as características dos aminoácidos presentes na sequência FASTA para detectar regiões hidrofóbicas típicas de domínios transmembranares (Krogh et al., 2001).
Construção do Modelo Tridimensional e Docagem Molecular
Os modelos tridimensionais da proteína SBP do transportador ABC foram gerados pelo programa ColabFold (Mirdita et al., 2022). Dos cinco modelos gerados, foi selecionado aquele de melhor qualidade com base em métricas estruturais, como IDDT (Integrated Distance Difference – Diferença de Distância Integrada), PAE (Predicted Aligned Error – Erro de Alinhamento Previsto), além de escores de cobertura e identidade, garantindo a avaliação da precisão estrutural em níveis globais e locais. O modelo com maior escore foi utilizado para investigar interações da SBP com moléculas-alvo por meio da docagem molecular, uma técnica que prevê a orientação preferencial de um ligante ao interagir com uma proteína, permitindo a análise da afinidade de ligação e da energia livre associada (Guedes et al., 2021).
A docagem molecular foi conduzida no programa DockThor (Guedes et al., 2021), sendo que tanto o modelo quanto os ligantes foram previamente preparados com adição de hidrogênio, remoção de moléculas de água não essenciais e protonação. Essas etapas foram realizadas utilizando os programas PDB2PQR (Unni et al., 2011) para a proteína e OpenBabel (O’Boyle et al., 2011) para os ligantes. Os resultados da docagem foram analisados em termos de energia livre de ligação (ΔG), identificando os complexos mais estáveis. Além disso, os resíduos de aminoácidos envolvidos nas interações entre os ligantes e o bolsão de ligação da proteína foram mapeados para uma melhor compreensão das interações moleculares.
Os dados obtidos foram validados por meio de comparações com informações experimentais disponíveis na literatura, e as estruturas dos complexos proteína-ligante foram visualizadas com ferramentas de modelagem molecular, permitindo uma análise detalhada dos resultados. Essa abordagem metodológica visa fornecer uma visão abrangente das interações da SBP com potenciais inibidores, contribuindo para o desenvolvimento de estratégias eficazes no combate à resistência bacteriana.
RESULTADOS E DISCUSSÃO
S. aureus possui transportador ABC de fosfato completo com conservado em procariotos
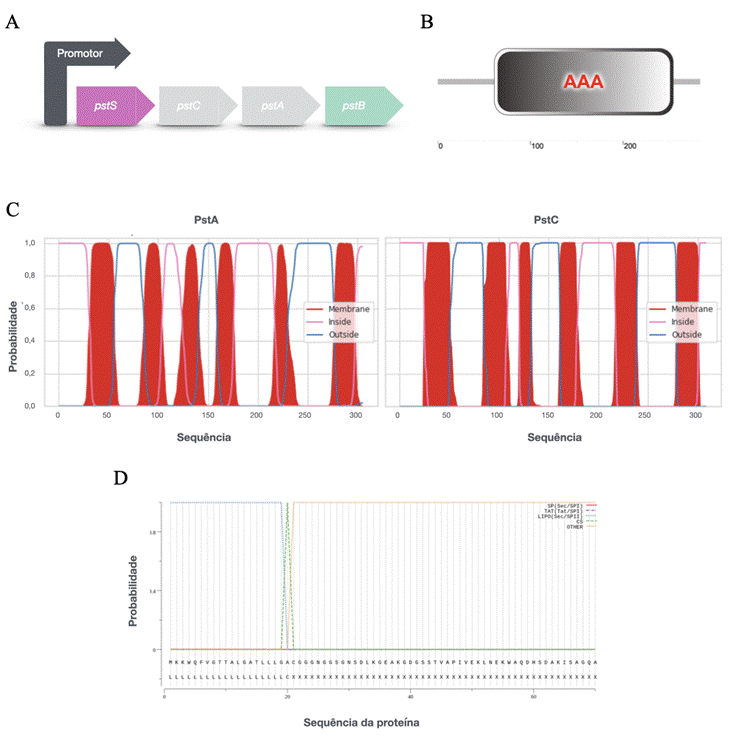
A análise do genoma de S. aureus no banco de dados Kegg permitiu a identificação de um possível transportador ABC de ligação ao fosfato, composto pelos genes pstS, pstC, pstA e pstB. Esses genes estão organizados em um óperon, regulado por um promotor localizado à montante do gene pstS (Figura 1A). Essa organização é comumente observada em bactérias, uma vez que um óperon corresponde a um conjunto de genes adjacentes no genoma de um organismo que são transcritos simultaneamente em uma única mensagem poligênica. Geralmente, os genes pertencentes ao mesmo óperon possuem funções correlacionadas ou participam de vias metabólicas semelhantes (Tjaden, 2019), como verificado no óperon pstSCAB de S. aureus.
A análise de domínios Pfam revelou a presença do domínio AAA na proteína PstB, indicando sua participação na família de proteínas ligadoras de nucleotídeos e caracterizando-a como uma provável ATPase (Figura 1B). Já os resultados obtidos por meio do programa DeepTMHMM sugeriram que as proteínas PstA e PstC possuem, cada uma, seis regiões transmembranares (Figura 1C), o que as classifica como possíveis permeases. Além disso, a proteína PstS, identificada como uma SBP, apresentou alta probabilidade de possuir um peptídeo sinal na região N-terminal, indicando que se trata de uma proteína secretada. Esse achado é compatível com a localização periplasmática típica das SBPs (Figura 1D).
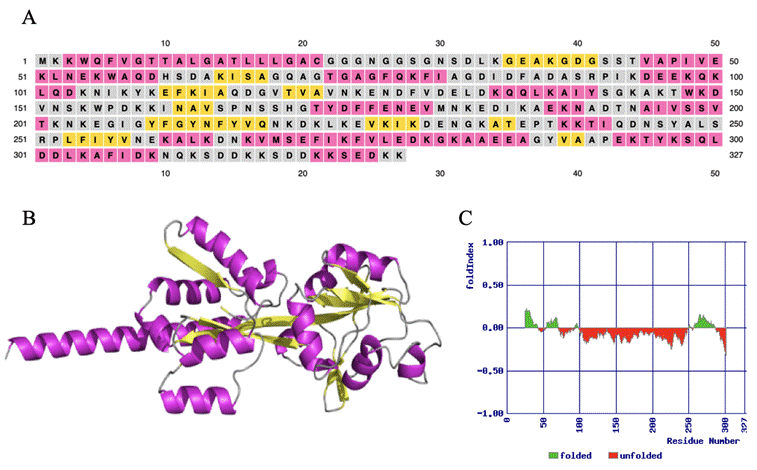
A predição da estrutura secundária da proteína PstS, realizada pelo Psipred, indicou a presença de regiões compostas por hélices-⍺, fitas-ꞵ e coil (Figura 2A). Essa conformação também foi observada no modelo tridimensional gerado pelo ColabFold (Figura 2B). Além disso, a análise conduzida pelo FoldIndex revelou a existência de regiões de desordem na estrutura da proteína (Figura 2C). O modelo estrutural obtido se assemelha ao padrão geral de uma SBP, que comumente apresenta entre duas e quatro fitas-ꞵ centrais flanqueadas por hélices-⍺. Na estrutura tridimensional, essa organização permite que a proteína se dobre ao redor do substrato durante a sua ligação (Scheepers; Lycklama a Nijeholt; Poolman, 2016).
A presença de regiões de desordem, apesar de indicar certa instabilidade estrutural, contribui para a flexibilidade da proteína, favorecendo a formação da interface de ligação e interação com o substrato (Mollica et al., 2016). O modelo de melhor qualidade gerado pelo ColabFold revelou uma estrutura composta por dois domínios formados predominantemente por hélices-⍺, unidos por duas fitas-ꞵ centrais. Essa conformação resulta na formação de uma cavidade entre os dois domínios, que funciona como bolsão de interação com o ligante, um padrão estrutural já descrito para proteínas SBP do tipo II (Locher, 2016).
Figura 1: Organização e Caracterização do óperon pstSCAB. A: Representação do operon pstSCAB, no qual os genes pstS, pstC, pstA e pstB codificam, as proteínas PstB, PstA, PstC e PstS, respectivamente. B: Esquema que mostra a presença da sequência do domínio AAA em proteínas que se ligam a nucleotídeos, indicando que a proteína PstB atua como uma ATPase em seu transportador. C: Análise do domínio transmembrana utilizando o programa DeepTMHMM, onde vermelho indica a região transmembrana das proteínas. A linha rosa indica os aminoácidos no citoplasma, enquanto a linha azul representa as regiões fora da membrana. D: Análise do peptídeo sinal da proteína ligadora de substrato. A linha azul representa peptídeo sinal, a linha verde representa o ponto de clivagem e em amarelo indica outras características da sequência que não sejam peptídeo sinal. Há probabilidade estimada de 99,89% de presença de peptídeo sinal entre os aminoácidos 1 e 20 da sequência.
Fonte: Autoria própria
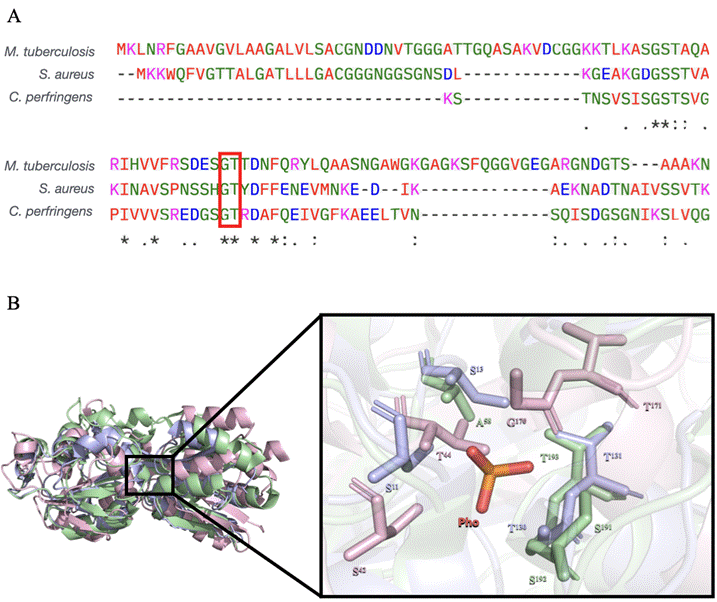
Pesquisas realizadas no banco de dados PDB revelaram proteínas transportadoras de fosfato de estrutura previamente resolvida, oriundas das bactérias Mycobacterium tuberculosis (PDB ID: 4LVQ) e Clostridium perfringens (PDB ID: 4Q8R), com identidades de sequência de 20,13% e 29,22%, respectivamente, em relação à proteína PstS de Staphylococcus aureus. Estudos estruturais anteriores indicaram que os aminoácidos presentes na SBP de M. tuberculosis, que interagem diretamente com o fosfato, são: S191, T193, G192 e A58 (Ferraris et al., 2014), enquanto em C. perfringens, os aminoácidos que se ligam ao fosfato são: S11, G130, T 131 e S13 (Gonzalez et al., 2014).
A partir do alinhamento múltiplo de sequências das SBPs dessas bactérias (Figura 3A), foi possível identificar a região específica de ligação ao fosfato em S. aureus, além dos aminoácidos conservados nessa interação: S42, G170 e T171. Além da conservação das sequências, os aminoácidos envolvidos na ligação ao fosfato mantêm a sua localização na estrutura tridimensional (Figura 3B). Esses achados foram confirmados pelos valores de RMSD (Root Mean Square Deviation - Raiz Quadrada do Desvio Quadrático Médio) entre as proteínas PstS de S. aureus e as de M. tuberculosis e C. perfringens, que foram de 5,82 Å e 4,65 Å, respectivamente.
Figura 2 - Caracterização estrutural da proteína PstS. A: Predição da estrutura secundária da proteína de acordo com a sequência FASTA. A cor rosa representa hélices-⍺, o amarelo representa fitas-ꞵ e o cinza representa coil. B: modelo tridimensional da proteína PstS, a cor rosa representa hélices-⍺, a cor amarela representa fitas-ꞵ e a cor cinza representa coil C: Gráfico gerado pelo Foldindex; cor verde representa regiões ordenadas e cor vermelha representa regiões desordenadas, revelando que a maior parte da proteína possivelmente está desordenada.
Fonte: Autoria própria
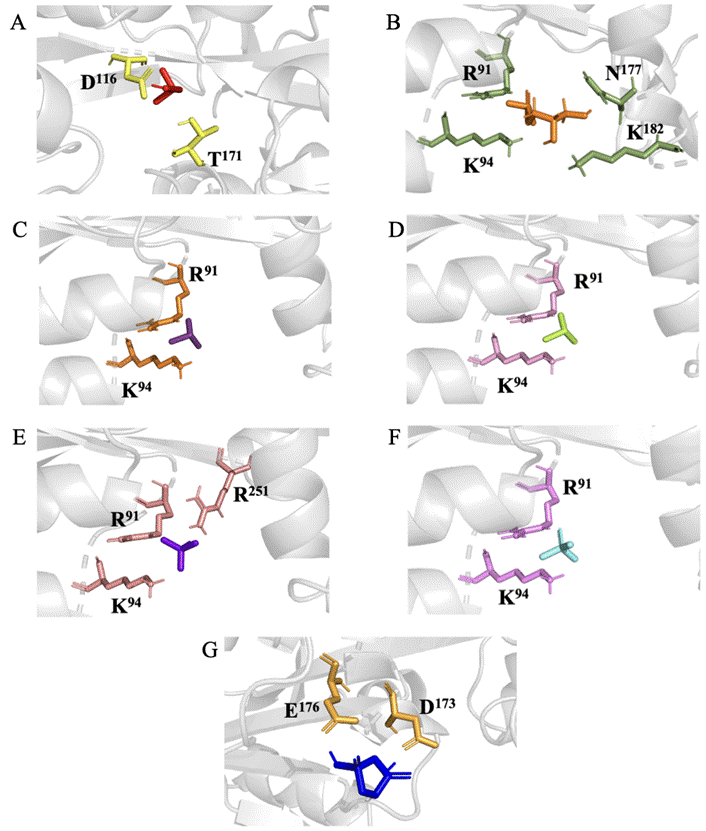
Apesar dos resultados obtidos no alinhamento das sequências, a docagem molecular sugeriu que a interação do fosfato é mais provável com os aminoácidos D116 e T171, localizados na região do bolsão de ligação da PstS (Figura 4A). O aspartato (D) e a treonina (T) podem se ligar ao fosfato por meio de ligações de hidrogênio, sendo que a treonina proporciona uma interação mais estável devido às forças eletrostáticas adicionais. A presença do grupo hidroxila na treonina permite a formação de uma rede de hidrogênio mais robusta com os oxigênios do fosfato, o que favorece uma ligação mais estável em comparação com o aspartato (Nishi; Shaytan; Panchenko, 2014). O programa DockThor utiliza diversos algoritmos que fornecem detalhes atômicos das interações entre o ligante e o sítio de ligação da proteína, além de calcular a energia livre, quantificando a qualidade da interação (Santos et al., 2020). A energia livre gerada na docagem foi de -5,989 kcal/mol, sugerindo que a interação entre a PstS e o fosfato ocorre de forma natural, com uma afinidade que é indicada pelo valor negativo da energia (Wallqvist; Covell, 1996).
Figura 3 - Identificação de aminoácidos ligantes na estrutura da PstS de S. aureus. A: Alinhamento múltiplo de proteínas transportadoras de fosfato de M. tuberculosis, S. aureus e C. perfringens; os aminoácidos conservados que possivelmente interagem especificamente com o fosfato estão circulados em vermelho. B: Modelo tridimensional da PstS de S. aureus (rosa), alinhada com proteínas ligadoras de fosfato de M. tuberculosis (verde) e de Clostridium perfringens (azul), destacando o bolsão de ligação e os respectivos aminoácidos que os compõem. O substrato fosfato está representado em vermelho, nomeado como “Pho”.
 Fonte: Autoria
própria
Fonte: Autoria
própria
Diferentes compostos ligados a fosfato podem interagir com PstS de S aureus
Um levantamento bibliográfico indicou que treze substâncias similares ao fosfato podem interagir com o sistema de transporte dessa molécula. Dentre as propriedades dessas moléculas, destacam-se a semelhança entre fosfonato, fosfito e ácido fosfônico, que exibem estrutura muito semelhante ao fosfato (Labotka; Omachi, 1987; Sevraim et al., 2017), a ação antibacteriana das moléculas de perclorato, monofluorofosfato, ácido fosfonoacético e fosfato de prata (Chudobova et al., 2013; Demonte et al., 2017; Pahontu et al., 2015; Shigi, 1989), a capacidade do foscarneto e do nitrato de interferir e inibir a captação de fosfato em organismos (Barak; van Rijn, 2000; Loghman-adham, 1996), e a semelhança estrutural entre compostos fosfatados e as moléculas de ácido pirofosfórico, carbamoil fosfato, sulfato e glicerofosfato (Gilmour, 2019; Lima et al., 2022; Ter-Ovanessian et al., 2021; Shinitzky et al., 2001).
A docagem molecular realizada entre esses ligantes e a proteína PstS de S. aureus sugere fortemente uma afinidade entre estes e o bolsão de ligação da proteína, com valores significativos de energia livre variando de -6,307 kcal/mol a -5,769 kcal/mol (Tabela 1). Observa-se que o glicerofosfato possui o menor valor de energia livre entre os ligantes, sugerindo uma maior afinidade pela proteína. Esse resultado ocorre possivelmente porque o glicerofosfato contém um grupo fosfato em sua composição e estudos anteriores já demonstraram que ele se liga diretamente de forma competitiva a proteínas ligadoras de fosfato de outras bactérias (Popova et al., 2010).
Tabela 1 - Relação entre os ligantes e a respectiva energia livre gerada na Docagem Molecular.
|
Ligantes |
Valor de energia livre (kcal/mol) |
|
Glicerofosfato |
-6.307 |
|
Fosfonato |
-6.107 |
|
Fosfito |
-6.090 |
|
Monofluorofosfato |
-6.085 |
|
Sulfato |
-6.077 |
|
Ácido Pirofosfórico |
-6.027 |
|
Carbamoil Fosfato |
-5.897 |
|
Nitrato |
-5.865 |
|
Ácido Fosfonocético |
-5.842 |
|
Foscarneto |
-5.841 |
|
Fosfato de Prata |
-5.833 |
|
Perclorato |
-5.827 |
|
Ácido Fosfônico |
-5.769 |
Fonte: Autoria própria
Segundo as análises de interação entre o sítio de ligação da proteína e os ligantes analisados, observou-se que algumas moléculas compartilham aminoácidos de interação e apresentam maior probabilidade de ligação espontânea, evidenciada por valores de energia livre mais negativos, em torno de -6,000 kcal/mol (Figura 4). O glicerofosfato, fosfonato, fosfito, monofluorofosfato e sulfato possivelmente compartilham os aminoácidos R91 e K94 no sítio de ligação da PstS de S. aureus, corroborando com o estudo de Gao Skolnick (2013), que mostrou a afinidade de moléculas similares pelos mesmos bolsões de ligação de proteínas SBP. Além dessas moléculas, a docagem molecular sugere que aquelas com energia livre acima de -5,769 kcal/mol também permanecem na região de ligação da PstS, como mostra a Figura 5.
Analisando o bolsão da proteína, nota-se que, apesar da semelhança estrutural, alguns ligantes podem interagir com aminoácidos diferentes enquanto permanecem no sítio de ligação. Segundo Gaudreault et al. (2012), a flexibilidade das proteínas permite que diferentes ligantes se acomodem e interajam com variados aminoácidos do sítio de ligação, o que corrobora com resultados semelhantes da docagem molecular realizada.
Figura 4 - Docagem molecular do fosfato e possíveis ligantes da PstS com energia livre menor que -6.000 kcal/mol A: Interação entre o fosfato representado em vermelho com os aminoácidos D116 e T171 representados em amarelo. B: Interação entre o Glicerofosfato representado em laranja com os aminoácidos R91, K94, N177 e K182 representados em verde. C: Interação entre o Fosfonato representado em roxo com os aminoácidos R91 e K94 representados em laranja. D: Interação entre o Fosfito representado em verde claro com os aminoácidos R91 e K94 representados em lilás. E: Interação entre o Monofluorofosfato representado em violeta com os aminoácidos R91, K94 e R251 representados em rosa. F: Interação entre o Sulfato representado em ciano e os aminoácidos R91 e K94 representados em lilás. G: Interação entre o Ácido Pirofosfórico representado em azul escuro e os aminoácidos E176 e D173 representados em amarelo mostarda.
Fonte: Autoria própria
Figura 5- Docagem molecular de possíveis ligantes da PstS com energia livre maior que -5.890kcal/mol A: Interação entre o Carbamoil fosfato representado em amarelo claro com os aminoácidos S250, R251 e E178 representados em vermelho escuro. B: Interação entre o Nitrato representado em laranja com os aminoácidos R91 e R251 representados em azul escuro. C: interação entre Ácido Fosfonoacético representado em verde claro com os aminoácidos K134, E176, N181 e E230 representados em magenta. D: Interação entre o Foscarneto representado em ciano com os aminoácidos R251 e E178 representados em verde oliva. E: Interação entre o Fosfato de prata representado em vermelho com os aminoácidos R91, K94 e R251 representados em cinza escuro. F:Interação entre o Perclorato representado em turquesa com os aminoácidos T72, G72, R91 e N177 representados em marrom. G: Interação entre o Ácido Fosfônico representado em rosa com os aminoácidos D193 e E196 representados em verde oliva.
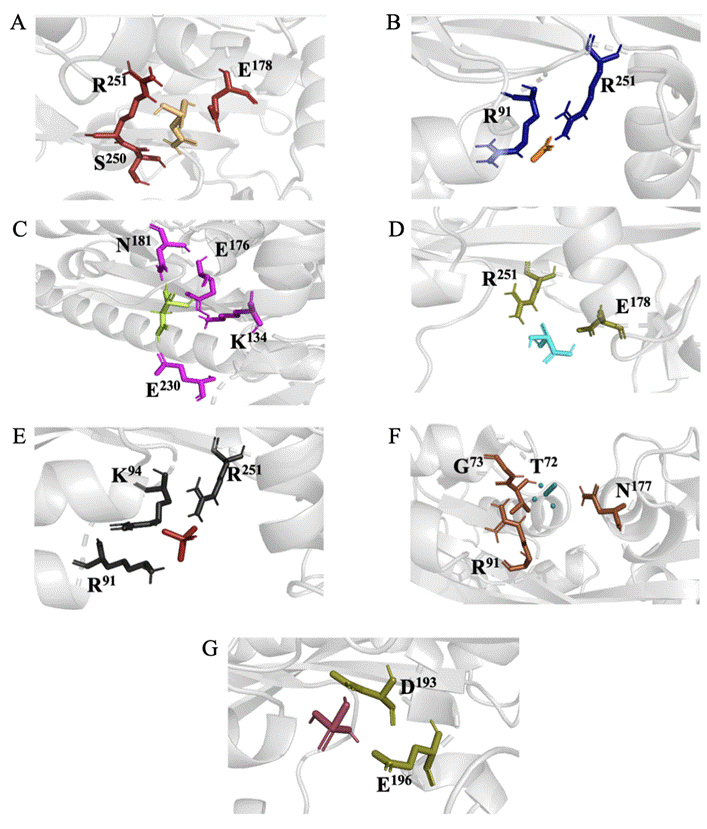
Fonte: Autoria própria
Devido à sua capacidade de prever a interação de ligantes de pequenas moléculas e proteínas, a docagem molecular está entre as estratégias mais frequentemente usadas no desenho racional de fármacos baseado em estrutura, contribuindo assim para a análise de processos bioquímicos fundamentais (Arya; Kaur, 2022). Estudos como o de Silva et al. (2023) enfatizam a importância da técnica para a estruturação e caracterização de transportadores ABC bacterianos específicos. Pesquisas como a de Zhang et al. (2022) corroboram com a utilização da docagem molecular para investigação e análise de interações com possíveis ligantes de transportadores ABC. Portanto, a utilização da técnica para investigar transportadores ABC e possíveis inibidores é considerada válida.
CONCLUSÃO
As análises computacionais indicam que Staphylococcus aureus possui quatro genes (pstS, pstC, pstA e pstB) organizados em um óperon que compõe o sistema de transporte ABC para captação de fosfato. A proteína PstS, codificada pelo gene pstS, atua como proteína ligadora de substrato (SBP) e apresenta interação espontânea com o fosfato, sendo os resíduos D116 e T171 os principais envolvidos no reconhecimento e ligação do íon.
A docagem molecular revelou que as moléculas glicerofosfato, fosfonato, fosfito, monofluorofosfato, sulfato, ácido pirofosfórico, carbamoil fosfato, nitrato, ácido fosfonoacético, foscarneto, fosfato de prata, perclorato e ácido fosfônico são potenciais ligantes da proteína PstS. Essas interações podem interferir no transporte de fosfato, um nutriente essencial para a sobrevivência e virulência de S. aureus, sugerindo um possível impacto na sua atividade metabólica e fisiológica.
Assim, este estudo contribui para a compreensão dos mecanismos de captação de fosfato em S. aureus e abre novas perspectivas para o desenvolvimento de estratégias terapêuticas voltadas à inibição do sistema de transporte ABC. A exploração de moléculas capazes de competir com o fosfato no sítio de ligação da PstS pode representar uma abordagem inovadora para o controle de infecções causadas por essa bactéria, especialmente em um cenário de crescente resistência antimicrobiana.
REFERÊNCIAS
AGUENA, M. Análise transcricional do operon pst de Escherichia coli. Tese (Doutorado em Microbiologia) —Instituto de Ciências Biomédicas da Universidade de São Paulo. São Paulo, p. 25. 2007. DOI: https://doi.org/10.11606/T.42.2007.tde-30012008-094907
ARMENTEROS, José Juan Almagro et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Naturebiotechnology, v. 37, n. 4, p.420-423, 2019. DOI: https://doi.org/10.1038/s41587-019-0036-z
ARYA, N.; KAUR, DR. A. Molecular Docking: A Review Paper. International Journal of Innovative Research in Engineering & Management, p. 140–146, 1 fev. 2022. DOI: https://doi.org/10.55524/ijirem.2022.9.1.25
BARAK, Y.; VAN RIJN, J. Relationship between Nitrite Reduction and Active Phosphate Uptake in the Phosphate-Accumulating Denitrifier Pseudomonas sp. Strain JR 12. Applied and Environmental Microbiology, v. 66, n. 12, p. 5236–5240, dez. 2000. DOI: https://doi.org/10.1128/aem.66.12.5236-5240.2000
CASSAT, J. E.; SKAAR, E. P. Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Seminars in Immunopathology, v. 34, n. 2, p. 215–235, 3 nov. 2011. DOI: https://doi.org/10.1007/s00281-011-0294-4
CHEUNG, G. Y. C.; BAE, J. S.; OTTO, M. Pathogenicity and virulence of Staphylococcus a u r e u s. Virulence, v. 1 2, n. 1, p. 5 4 7 – 5 6 9, 3 1 j a n. 2 0 2 1. D O I: https://doi.org/10.1080/21505594.2021.1878688
CHUDOBOVA, D. et al. Comparison of the effects of silver phosphate and selenium nanoparticles on Staphylococcus aureus growth reveals potential for selenium particles to prevent infection. FEMS Microbiology Letters, v. 351, n. 2, p. 195–201, 30 dez. 2013. DOI: https://doi.org/10.1111/1574-6968.12353
DEMONTE, A. M.; DIEZ A. D. M.; NALEWAY C.; IGLESIAS A. A.; BALLICORA M. A. Monofluorophosphate Blocks Internal Polysaccharide Synthesis in Streptococcus mutans. PLOS ONE, v. 12, n. 1, p. e0170483, 26 jan. 2017. DOI: https://doi.org/10.1371/journal.pone.0170483
FERRARIS, D. M.; SPALLEK R.; OEHLMANN W.; SIGH M.; RIZZI M. Crystal structure of the Mycobacterium tuberculosis phosphate binding protein PstS3. Proteins: Structure, Function, and Bioinformatics, v. 82, n. 9, p. 2268–2274, 24 mar. 2014. DOI: https://doi.org/10.1002/prot.24548
GAO, M.; SKOLNICK, J. A Comprehensive Survey of Small-Molecule Binding Pockets in Proteins. PLoS Computational Biology, v. 9, n. 10, p. e1003302, 24 out. 2013. DOI: https://doi.org/10.1371/journal.pcbi.1003302
GAUDREAULT, F.; CHARTIER, M.; NAJMANOVICH, R. Side-chain rotamer changes upon ligand binding: common, crucial, correlate with entropy and rearrange hydrogen bonding. Bioinformatics, v. 28, n. 18, p. i423–i430, 7 set. 2012. DOI: https://doi.org/10.1093/bioinformatics/bts395
GILMOUR, R. B. Phosphoric Acids and Phosphates. Kirk-Othmer Encyclopedia of Chemical Technology, p. 1–51, 12 jun. 2019. DOI: https://doi.org/10.1002/0471238961.1608151907011804.a01.pub3
GONZALEZ, D.; RICHEZ M.; BERGONZI C.; CHABRIERE E.; ELIAS M. Crystal structure of the phosphate-binding protein (PBP-1) of an ABC type phosphate transporter from Clostridium perfringens. Scientific Reports, v. 4, n. 1, 16 out. 2014. DOI: https://doi.org/10.1038/srep06636
GUEDES, I. A. et al. New machine learning and physics-based scoring functions for drug discovery. Scientific Reports, v. 11, n. 1, 4 fev. 2021. DOI: https://doi.org/10.1038/s41598-021-82410-1
HALLGREN, Jeppe et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. BioRxiv, 2022. DOI: https://doi.org/10.1101/2022.04.08.487609
HOLLENSTEIN, K.; DAWSON, R. J.; LOCHER, K. P. Structure and mechanism of ABC transporter proteins. Current Opinion in Structural Biology, v. 17, n. 4, p. 412–418, ago. 2007. DOI: https://doi.org/10.1016/j.sbi.2007.07.003
HORSMAN, G. P.; ZECHEL, D. L. Phosphonate Biochemistry. Chemical Reviews, v. 117, n. 8, p. 5704–5783, 27 out. 2016. DOI: https://doi.org/10.1021/acs.chemrev.6b00536
KELLIHER, J. L. et al. Staphylococcus aureus Preferentially Liberates Inorganic Phosphate from Organophosphates in Environments where This Nutrient Is Limiting. Journal of Bacteriology, v. 202, n. 22, 31 ago. 2020. DOI: https://doi.org/10.1128/jb.00264-20
KROGH, A.; LARSSON, B.; VON HEIJNE, G.; SONNHAMMER, E.L.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. Journalof Molecular Biology, v. 305, n.3, p. :567-580, 2001. DOI: https://doi.org/10.1006/jmbi.2000.4315
LABOTKA, R. J.; A OMACHI. Erythrocyte anion transport of phosphate analogs. Journal of biological chemistry/The Journal of biological chemistry, v. 262, n. 1, p. 305–311, 1 jan. 1987. DOI: https://doi.org/10.1016/S0021-9258(19)75927-4.
LARKIN M.A., BLACKSHIELDS G., BROWN N.P., CHENNA R., MCGETTIGAN P.A., MCWILLIAM H., VALENTIN F., WALLACE I.M., WILM A., LOPEZ R., THOMPSON J.D., GIBSON T.J. AND HIGGINS D.G. Clustal W and Clustal X version 2.0. bioinformatics, v. 23, n. 21, p. 2947-2948, 2007. DOI: https://doi.org/10.1093/bioinformatics/btm404
LETUNIC, Ivica; KHEDKAR, Supriya; BORK, Peer. SMART: recent updates, new developments and status in 2020. Nucleic acids research, v. 49, n. D1, p. D458-D460, 2021. https://doi.org/10.1093/nar/gkaa937
LEWIS, VG.; WEEN, MP.; MCDEVITT, CA. The role of ATP-binding cassette transporters in bacterial pathogenicity. Protoplasma, v. 249, p. 919-942, 2012. DOI: https://doi.org/10.1007/s00709-011-0360-8
LEWINSON, O.; LIVNAT-LEVANON, N. Mechanism of Action of ABC Importers: Conservation, Divergence, and Physiological Adaptations. Journal of Molecular Biology, v. 429, n. 5, p. 606–619, mar. 2017. DOI: https://doi.org/10.1016/j.jmb.2017.01.010
LIMA, M. A.; RUDD T. R.; FERNING D. G.; YATES E. A. Phosphorylation and sulfation share a common biosynthetic pathway, but extend biochemical and evolutionary diversity of biological macromolecules in distinct ways. Journal of The Royal Society Interface, v. 19, n. 193, ago. 2022. DOI: https://doi.org/10.1098/rsif.2022.0391
LOCHER, K. P. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nature Structural & Molecular Biology, v. 23, n. 6, p. 487–493, jun. 2016. DOI: 10.1038/ nsmb.3216 DOI: https://doi.org/10.1038/nsmb.3216
LOGHMAN-ADHAM, M. Use of phosphonocarboxylic acids as inhibitors of sodium phosphate cotransport. General Pharmacology: The Vascular System, v. 27, n. 2, p. 305– 312, mar. 1996. DOI: https://doi.org/10.1016/0306-3623(95)02017-9
MIRDITA, M.; SCHÜTZE K.; MORIWAKI Y.; HEO L.; OVCHINNIKOV S.; STEINEGGER M. ColabFold: making protein folding accessible to all. Nature Methods, v. 19, p. 1–4, 30 maio 2022. DOI: https://doi.org/10.1038/s41592-022-01488-1
MLYNARCZYK-BONIKOWSKA, B. KOWALEWSKI C.; KROLAK-ULINSKA A.; MARUSZA W. Molecular Mechanisms of Drug Resistance in Staphylococcus aureus. International Journal of Molecular Sciences, v. 23, n. 15, p. 8088, 1 jan. 2022. DOI: https://doi.org/10.3390/ijms23158088
MOLLICA, L.; BESSA L. M.; HANOULLE X.; JENSEN M. R.; BLACKLEDGE M.; SCHNEIDER R. Binding Mechanisms of Intrinsically Disordered Proteins: Theory, Simulation, and Experiment. Frontiers in Molecular Biosciences, v. 3, 9 set. 2016. DOI: https://doi.org/10.3389/fmolb.2016.00052
NISHI, H.; SHAYTAN, A.; PANCHENKO, A. R. Physicochemical mechanisms of protein regulation by phosphorylation. Frontiers in Genetics, v. 5, 7 ago. 2014. DOI: https://doi.org/10.3389/fgene.2014.00270
O'BOYLE, Noel M.; BANCK M.; JAMES C. A.; MORLEY C.; VANDERMEERSCH T.; HUTCHISON G. R. Open Babel: An open chemical toolbox. Journal of cheminformatics, v. 3, p. 1-14, 2011. DOI: https://doi.org/10.1186/1758-2946-3-33
PAHONȚU, E. et al. Synthesis, Characterization, Crystal Structure and Antimicrobial Activity of Copper (II) Complexes with the Schiff Base Derived from 2-Hydroxy-4-Methoxybenzaldehyde. Molecules, v. 20, n. 4, p. 5771–5792, 2 abr. 2015.DOI: https://doi.org/10.3390/molecules20045771
PEGOS, V. R. Nascimento J. F.; SOBREIRA T. J. P.; PAULETTI B. A.; PAES-LEME A. Phosphate regulated proteins of Xanthomonas citri subsp. citri: a proteomic approach. Journal of proteomics, v. 108, p. 78-88, 2014. DOI: https://doi.org/10.1016/j.jprot.2014.05.005
PEREIRA, CT.; ROESLER, C.; FARIA, JN.; FESSEL, MR.; BALAN, A. Sulfate-Binding Protein (Sbp) from Xanthomonas citri: Structure and Functional Insights. Molecular Plant Microbe Interactions, v.30, p. 578-588, 2017. DOI: https://doi.org/10.1094/mpmi-02-17-0032-r
POPOVA, Y. THAYUMANAVAN, P. LONATI, E. AGROCHAO,M. THEVELEN, J.M. Transport and signaling through the phosphate-binding site of the yeast Pho84 phosphate transceptor. Proceedings of the National Academy of Sciences, v. 107, n. 7, p. 2890–2895, 1 fev. 2010. DOI: https://doi.org/10.1073/pnas.0906546107
PRILUSKKY, J. FOLDER, CE. ZEEV-BEN-MORDEHAI, T. RYDBERG, EH. MAN, O. BECKMANN, JS. SILMAN, I. SUSSMAN, JL. FoldIndex©: a simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics, v. 21, n. 16, p. 3435-3438, 2005. DOI: https://doi.org/10.1093/bioinformatics/bti537
REECE, J. B., URRY, L. A., CAIN, M. L., WASSERMAN, S. A., MINORSKY, P. V. JACKSON, R. B. (2011). Membrane structure and function. In Campbell biology, 10th ed., p. 127. San Francisco, CA: Pearson.
REES, DC. JOHNSON, E. LEWINSON, O. ABC transporters: the power to change. Nature Reviews Molecular Cell Biology, v.10, p. 218-227, 2009. DOI: https://doi.org/10.1038/nrm2646
SALAMOV, A.; SOLOVYEVAND, V. Automatic annotation of microbial genomes and metagenomic sequences. In: LI, R.W. (ed.). Metagenomics and its applications in agriculture, biomedicine and environmental studies. New York: Nova Science Publishers, 2011. p. 61-78.
SAMPAIO, A.; PEGOS, VANESSA; OSHIRO, ELISA; BALAN, A. The periplasmic binding protein NrtT affects xantham gum production and pathogenesis in Xanthomona citri. FEBS Open Bio, v. 7, p. 1499-1514, 2017. DOI: https://doi.org/10.1002/2211-5463.12281
SANDOVAL, J.; SCOTTIE, E. Investigating the role of phosphate homeostasis in Staphylococcus aureus virulence. 2021. Tese (Mestrado em Microbiologia) - Universidade de Illinois Urbana-Champaign, Illinois
SANTOS, K. B. et al. Highly Flexible Ligand Docking: Benchmarking of the DockThor Program on the LEADS-PEP Protein–Peptide Data Set. Journal of Chemical Information and Modeling, v. 60, n. 2, p. 667–683, 10 jan. 2020. DOI: https://pubs.acs.org/doi/10.1021/acs.jcim.9b00905
SCHEEPERS, G. H.; LYCKLAMA A NIJEHOLT, J. A.; POOLMAN, B. An updated structural classification of substrate-binding proteins. FEBS Letters, v. 590, n. 23, p. 4393–4401, 23 out. 2016. DOI: https://doi.org/10.1002/1873-3468.12445
SEVRAIN, M. C.; BERCHEL, M.; COUTHON, H.; JAFFRÈS, Phosphonic acid: preparation and applications. Beilstein Journal of Organic Chemistry, v. 13, p. 2186–2213, 20 out. 2017. DOI: https://doi.org/10.3762/bjoc.13.219
SHIGI, Y. Inhibition of bacterial isoprenoid synthesis by fosmidomycin, a phosphonic acid-containing antibiotic. Journal of Antimicrobial Chemotherapy, v. 24, n. 2, p. 131–145, 1989. DOI: https://doi.org/10.1093/jac/24.2.131
SHINITZKY, M.; HAYMOVITZ R.; NEMAS M.; CAHANA N.; MAMILLAPALLI R.; SEGER R. Induction of intracellular signalling by cyclic glycerophosphates and their deoxy analogues. European Journal of Biochemistry, v. 267, n. 9, p. 2547–2554, 25 dez. 2001. DOI: https://doi.org/10.1046/j.1432-1327.2000.01265.x
SILVA, K.; PRAZERES R. A.; CURSELLI F.; CREMONESI A. S. Structural Characterization and Molecular Docking of Polyamine Transporters in Enterobacter Cloacae. SciMed Central, 16 maio 2023 DOI: https://doi.org/10.47739/2333-7109/1039
TANAKA, KJ.; SONG, S.; MASON, K.; PINKETT, HW. Selective substrate uptake: The role of ATP-binding cassette (ABC) importers in pathogenesis. Biochimica et Biophysica Acta (BBA) - Biomembranes, v. 1860, p.868-877, 2017. DOI: https://doi.org/10.1016/j.bbamem.2017.08.011
TER BEEK, Josy; GUSKOV, Albert; SLOTBOOM, Dirk Jan. Structural diversity of ABC transporters. Journal of General Physiology, v. 143, n. 4, p. 419-435, 2014. https://doi.org/ 10.1002/1873-3468.13935
TER-OVANESSIAN, L. M. P.; RIGAUD B.; MEZZETTI A.; LAMBERT J.; MAUREL M. Carbamoyl phosphate and its substitutes for the uracil synthesis in origins of life scenarios. Scientific Reports, v. 11, n. 1, 29 set. 2021. DOI: https://doi.org/10.1038/s41598-021-98747-6
TJADEN, B. A computational system for identifying operons based on RNA-seq data. Methods, v. 176, abr. 2019. DOI: https://doi.org/10.1016/j.ymeth.2019.03.026
UNNI, Samir et al. Web servers and services for electrostatics calculations with APBS and PDB2PQR. Journal of computational chemistry, v. 32, n. 7, p. 1488-1491, 2011. https:// doi.org/10.1002/jcc.21720
VON HEIJNE, G. The signal peptide. Journal of Membrane Biology, v. 115, p. 195-201, 1990. DOI: https://doi.org/10.1007/BF01868635
WALLQVIST A.; COVELL D. G. Docking enzyme-inhibitor complexes using a preference based free-energy surface. Proteins: Structure, Function, and Bioinformatics, v. 25, n. 4, p. 403–419, ago. 1996. DOI: https://doi.org/10.1002/(SICI)1097-0134(199608)25:4<403::AID-PROT1>3.0.CO;2-E
ZHANG, Y. et al. Discovery and structure of a widespread bacterial ABC transporter specific for ergothioneine. Nature Communications, v. 13, n. 1, 8 dez. 2022. DOI: https://doi.org/10.1038/s41467-022-35277-3